II黏多糖贮积症诊疗指南概述黏多糖贮积症(mucopolysaccharidosis,MPS)是一组复杂的、进行性多系统受累的溶酶体病,是由于降解糖胺聚糖(亦称酸性黏多糖,glycosaminoglycan,GAGs)的酶缺乏所致
不能完全降解的黏多糖在溶酶体中贮积,可造成面容异常、神经系统受累、骨骼畸形、肝脾增大、心脏病变、角膜混浊等
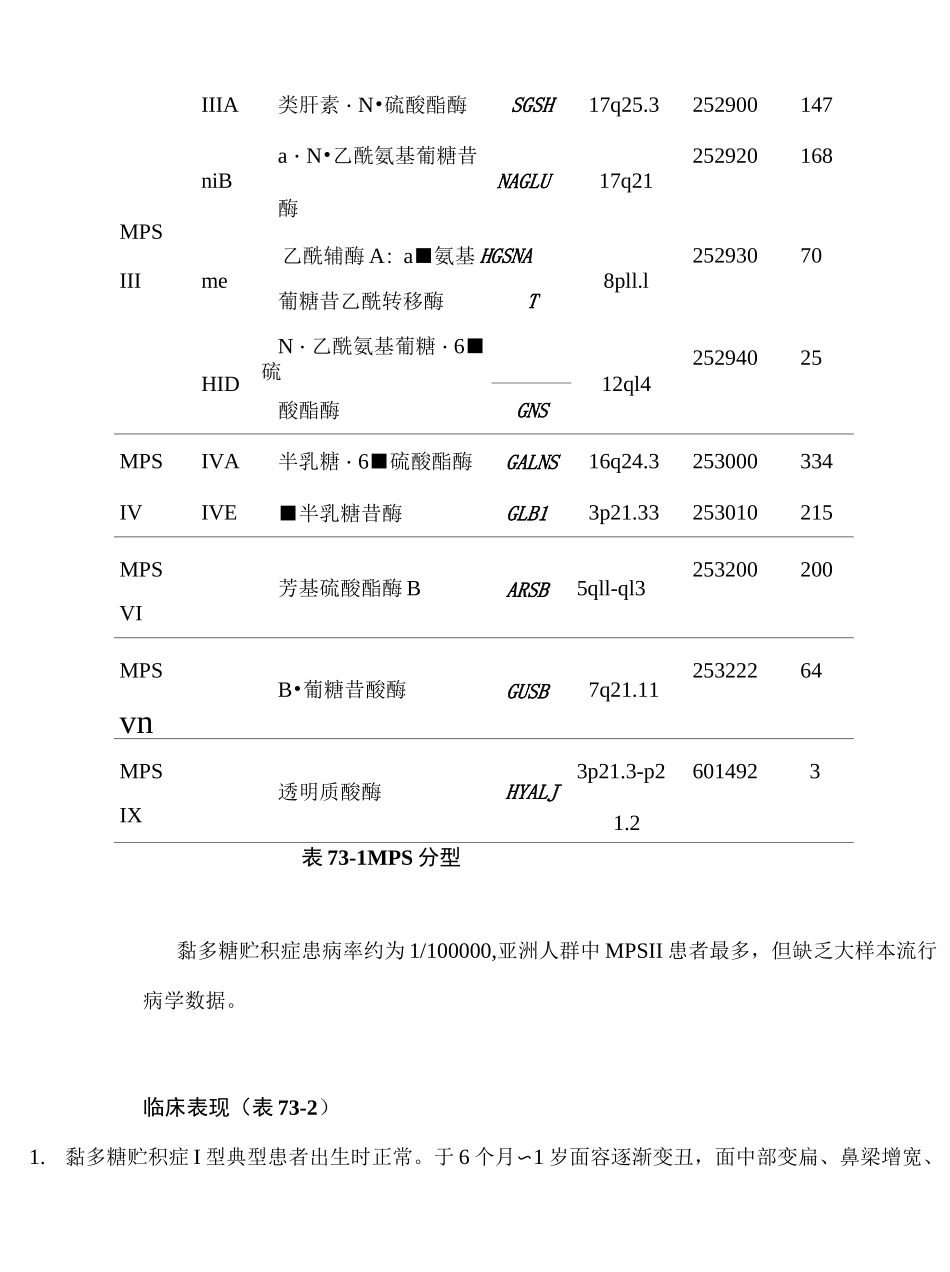
病因和流行病学黏多糖贮积症共分为7型(表73-1),涉及11个基因编码的11种溶酶体酶,除MPSII型为X连锁遗传外,其余皆属常染色体隐性遗传
所有黏多糖贮积症的酶学分析是该病诊断的金标准
MPS基因定位MIM已知突变分型亚型缺陷酶基因IH607014MPSISa・L•艾杜糖昔酶IDUA4pl6
3607016279IIH/I607015SMPS309900632艾杜糖醛酸硫酸酯酶IDSXq28IIIA类肝素・N•硫酸酯酶SGSH17q25
3252900147a・N•乙酰氨基葡糖昔252920168niB酶NAGLU17q21MPS乙酰辅酶A:a■氨基HGSNA25293070IIIme葡糖昔乙酰转移酶T8pll
lHIDN・乙酰氨基葡糖・6■硫12ql425294025酸酯酶GNSMPSIVA半乳糖・6■硫酸酯酶GALNS16q24
3253000334IVIVE■半乳糖昔酶GLB13p21
33253010215MPS芳基硫酸酯酶BARSB5qll-ql3253200200VIMPSB•葡糖昔酸酶GUSB7q21
1125322264vnMPS透明质酸酶HYALJ3p21
3-p26014923IX1
2表73-1MPS分型黏多糖贮积症患病率约为1/100000,亚洲人群中MPSII患者最多,但缺乏大样本流行病学数据
临床表现(表73-2)1
黏多糖贮积症I型典型患者出生时正常
于6个月〜1岁面容逐渐变丑,面中部变扁、鼻梁增宽、角膜混