2020 版:脊髓性肌萎缩症遗传学诊断专家共识(全文)脊髓性肌萎缩症(spinalmuscularatrophy,SMA)是儿童最常见的神经肌肉病,以脊髓前角 a-运动神经元退化变性导致的肌无力和肌萎缩为主要临床特征
本共识中 SMA 特指位于 5q13 的运动神经元存活基因 1(SMN1;OMIM600354)致病性变异所导致的 5q-SMA
SMA 发病率约为 1/10000,人群携带率约为 1/50[1]
2019 年中国大陆上市了疾病修正治疗药物诺西那生钠注射液,也相继发表了 SMA 多学科管理专家共识[2],标志着SMA 在我国进入了一个全新的精准诊治和管理时期
SMA 的携带者和新生儿筛查在一些国家和地区已常规开展[3,4],我国一些地区也逐渐开始筛查[5,6],SMA 预防窗口进一步提前
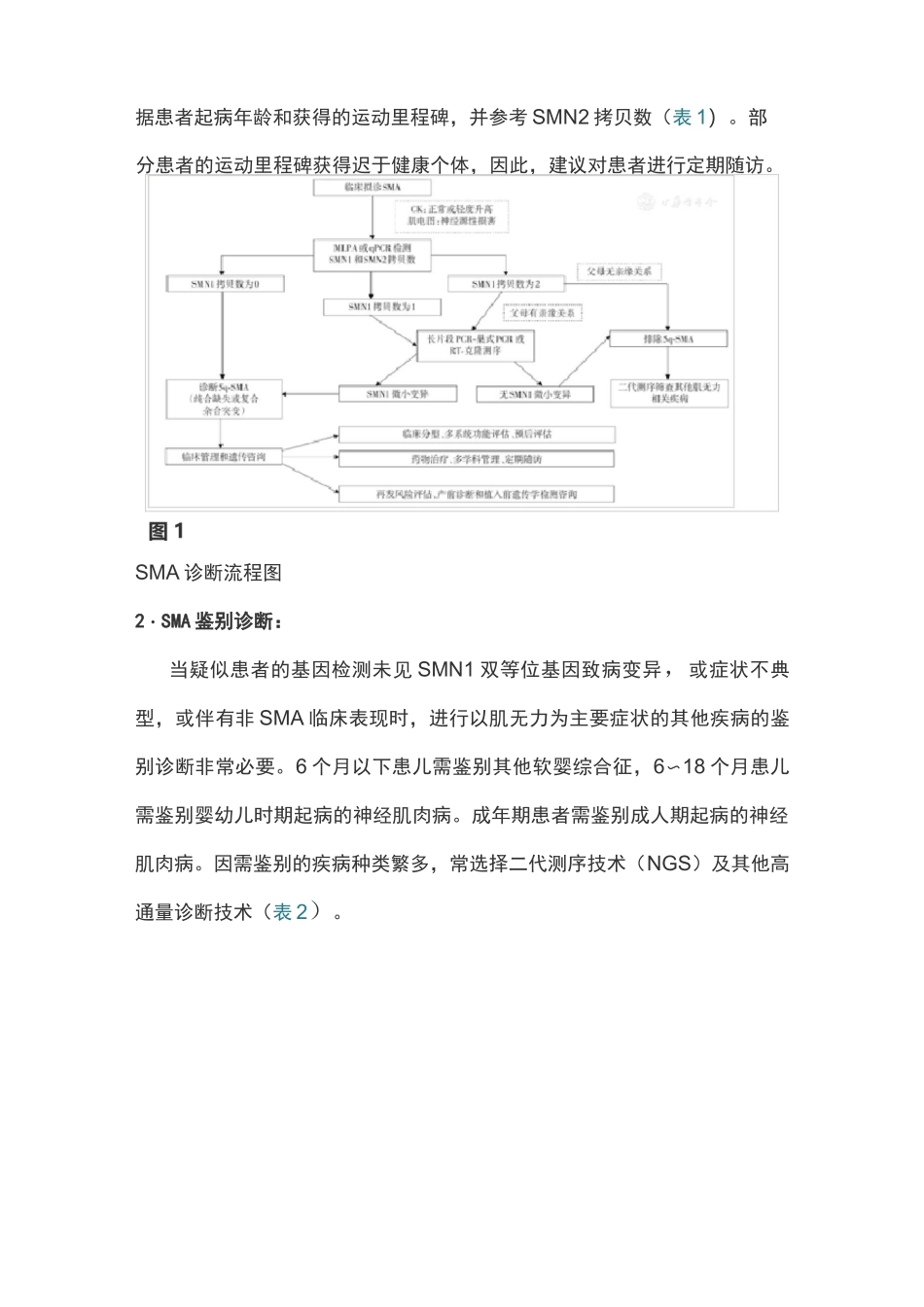
SMA 的致病基因 SMN1 和修饰基因 SMN2(OMIM601627) 高度同源,SMN1 决定疾病的发生,SMN2 影响疾病的严重程度和进展,使得SMA 的遗传学诊断不同于绝大多数单基因遗传病
规范 SMA 遗传学诊断及应用对于临床诊治、管理、预防和遗传咨询将提供重要帮助
本共识参照国内外近年 SMA 临床诊疗实践和指南共识[2,7,8,9,10],由具有实践经验的多学科专家研究起草,包括了患者和携带者基因型、基因诊断技术的适用性和局限性,以及基因诊断、产前诊断、植入前遗传学检测和携带者筛查的要点及遗传咨询等内容,并对 SMN2 拷贝数的临床价值提出了一些建议
旨在为医生和实验室人员的临床实践提供指导帮助
临床表现与分型SMA 患者起病年龄差异性大,从出生前至成人期均可发病
主要表现为以四肢近端为主的进行性肌无力和肌萎缩,随着疾病进展何出现呼吸、消化、骨骼等多系统受累
根据起病年龄、运动里程碑及病情进展程度,SMA 分为五型
近年的临床实践趋于将每型 SMA 进一步分为