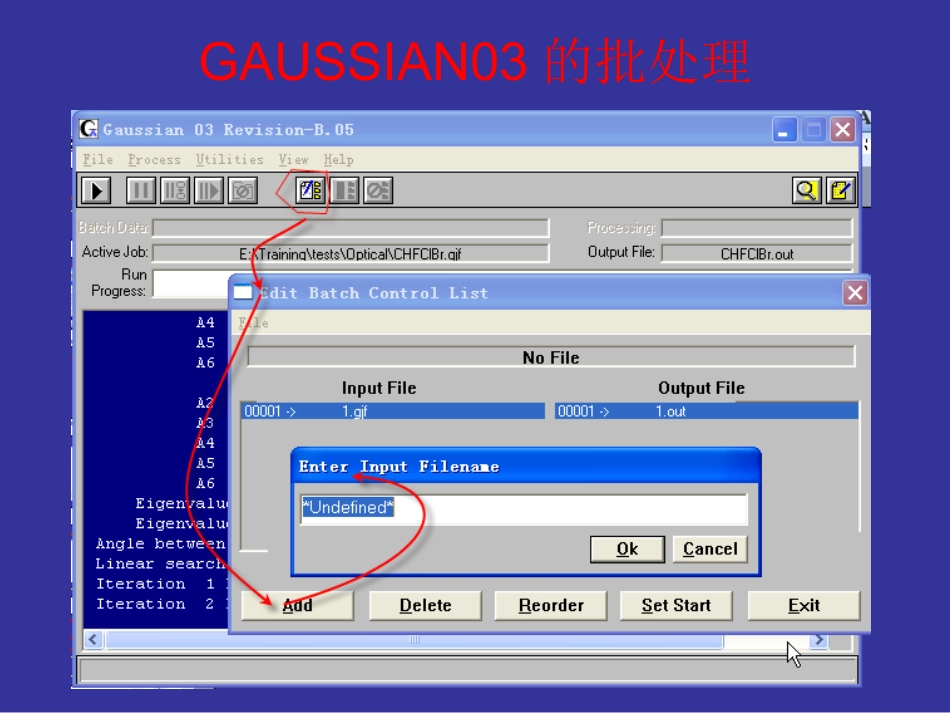
Gaussian培训张祥G03W的一些设置GAUSSIAN03的批处理Default
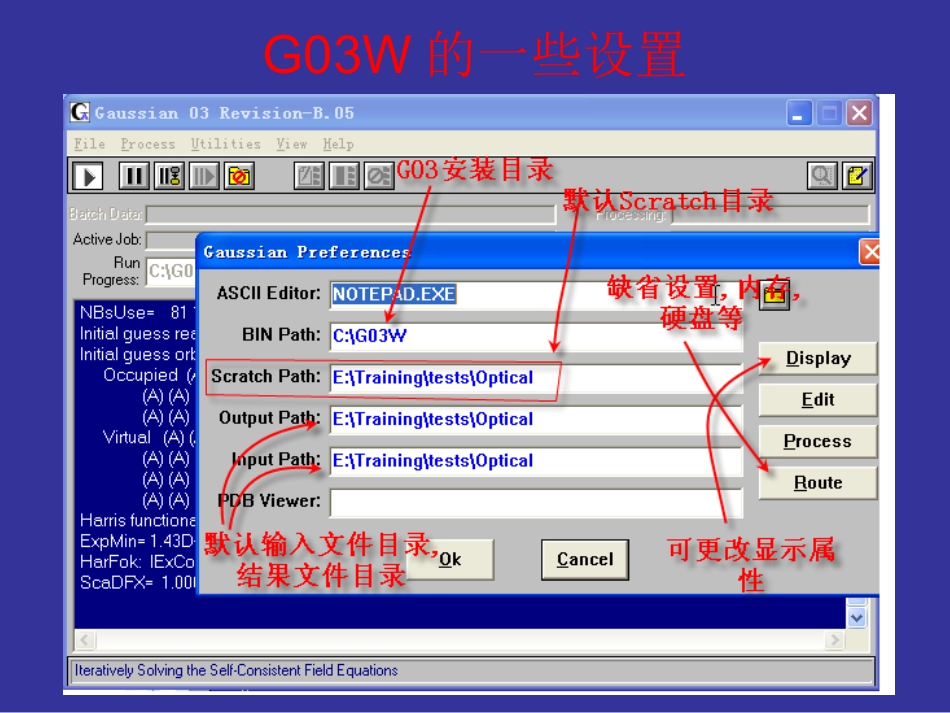
Rou设置•在Scratch文件夹中的Default
Rou文件中设置G03程序运行的省缺参数:•-M-200MW•-P-4•-#-MaxDisk=10GB•-#-SCF=ConventionalorDirect•-#-MP2=NoDirectorDirect•-#-OPTCYC=200•-#-SCFCYC=200•-#-IOPs设置如iop(2/16=1)Default
Rou设置中的冲突•Defaultroute:MaxDisk=2GBSCF=DirectMP2=DirectOPTCYC=200SCFcyc=100iop(2/16=1)iop(5/13=1)•------------------•#ccsd/6-31G**opt•------------------•L903/L905andL906canonlydoMP2
问题在于,MP2=Direct
去掉这个设置,CCSD的作业就能进行了
因此,建议在Default设置中只设置,内存,最大硬盘,等项
势能面上的局域极小点限制性空间轨道和非限制性空间轨道闭壳层缺省为限制性空间轨道计算;开壳层缺省为非限制性空间轨道计算应该用开窍层计算的体系•具有奇数个电子的体系(例如,离子或自由基)•激发态•具有特殊电子结构的体系,如外层具有2个或多个为成对电子的体系•具有键的解离,如反应中,需要将一对电子分开的过程的体系,这样的体系限制性计算将导致不正确结果(尽管电子数是偶数)•对于某些体系,需要用额外的关键词Guess=mix或者Guess=alter来强制得到非限制性的波函数
冻结核近似•FC=FREEZENCORE•相关能计算中不考虑内壳层轨道的贡献•要考虑内壳层轨道的贡献用FULL•指定相关能计算轨道,用RW或Window•FC,Full,