使用DMol3 中的离域内坐标对固体进行几何优化 DMol3 的面向分子的离域内坐标优化机制为大分子系统提供了一套良好的方案
在 MS Modeling 的DMol3 中,这个机制被扩展到周期性系统
这个基于离域内坐坐标的新型优化工具还有能力处理以下体系: z 高度坐标化系统,比如密堆积固体 z 片断系统,比如分子晶体,其中的内坐标并不是遍及整个优化空间 z 包含了在优化过程中的迪卡尔坐标限制 内部的有效工作表明,对周期性体系而言,这个状态图式的离域内坐标优化方案的效率要比迪卡尔坐标方法高出 2-5 倍,而迪卡尔坐标方法是先在进行固态计算的标准方法
在这个指南中,我们将利用DMol3 的优化工具,使用离域内坐标方法对一分子筛结构进行几何优化
演示包括以下步骤: z 建立一个计算 DMol3 任务 z 控制工作设置并开始计算 z 使用服务器控制台控制计算任务 z 检验计算结果 1
建立一个计算 DMol3 任务 第一步是输入需要进行优化的分子筛结构
MS Modeling 提供了现成的比较广泛的分子筛结构
本例中,我们将优化菱沸石(chabazite)分子筛
点击工具栏里的Import 按钮
找到 Examples\Documents\3D Model\CHA
xsd 并点击输入文件对话栏上的Import 按钮
在 3D 浏览器中右击鼠标,选择 Display Style,把显示方式改为Polyhedron
用Polyhedra 方式显示的CHA 的3D 图
点 击 工 具 栏 上 的DMol3 按 钮 选 择 Calculation , 或 者 从 菜 单 栏 里 选 择Module|DMol3|Calculation
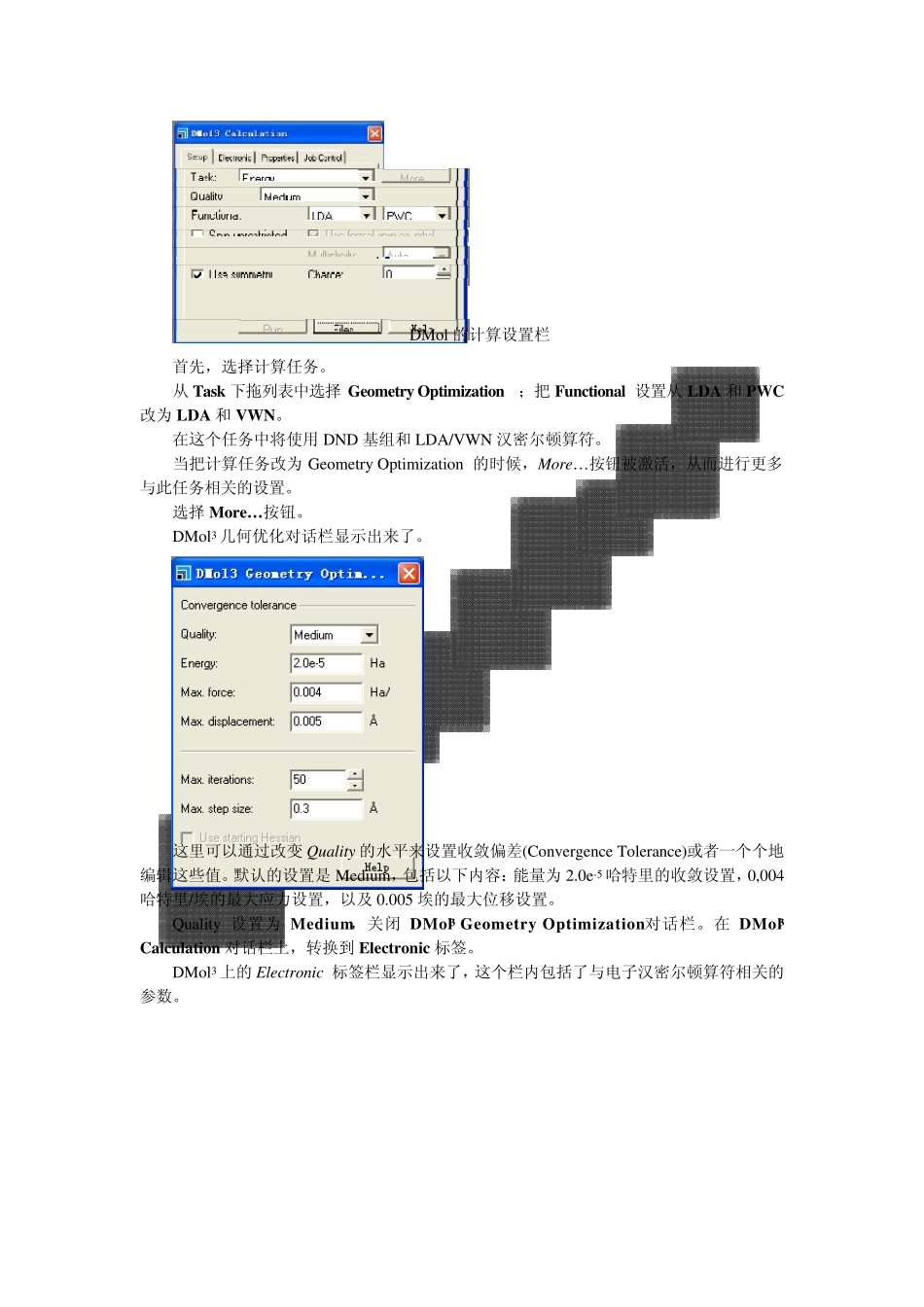
DMol 的计算设置栏 首先,选择计算任务
从 Task 下拖列表中选择Geometry Optimization;把 Fu