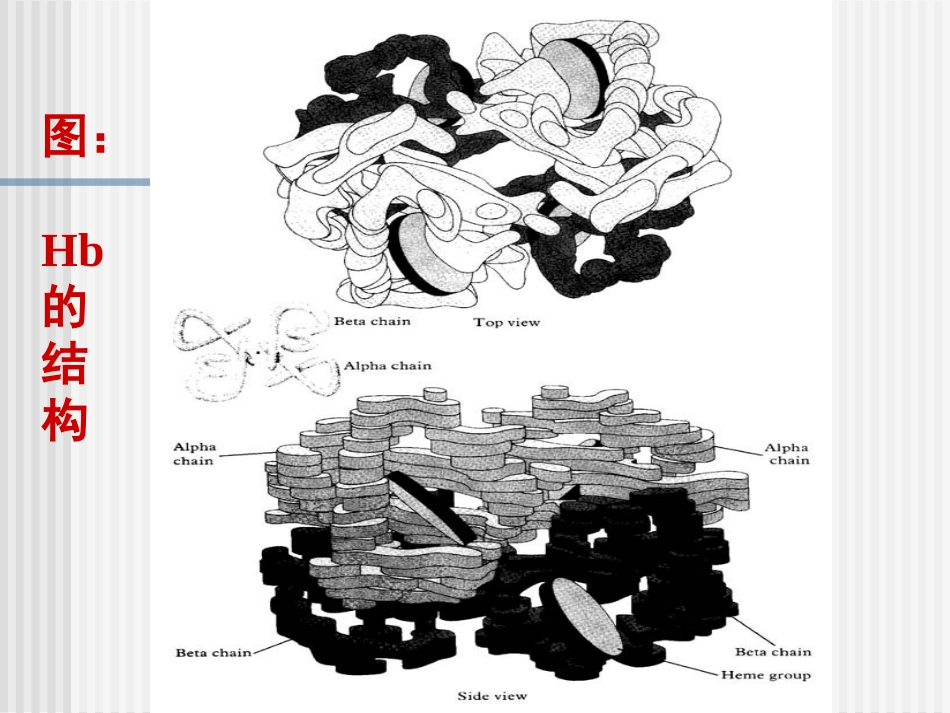
第八章生化遗传病第八章生化遗传病分子病:由于蛋白质分子结构或合成量异常所引起的疾病第一节血红蛋白病一.正常Hb的分子结构及其遗传控制血红蛋白(Hemoglobin—Hb)1.Hb的分子结构结合蛋白:血红素+珠蛋白(Heme)(globin)一条肽链+一分子血红素→Hb单体4个Hb单体→球形四聚体(Hb)图:Hb的结构图:Hb的四聚体结构2
Hb种类珠蛋白链(6种):、、、、、分两大类:链(类链):、----141个氨基酸组成β链(类β链):、、、(G、A)-----146个氨基酸组成Hb类型:成人Hb:HbA2297~98%HbA2222~3%HbF<1%胎儿Hb:HbF22胚胎Hb:HbGowerⅠ22(妊娠12周内)HbGowerⅡ22HbPortland223
人类珠蛋白基因结构和表达类珠蛋白基因簇类珠蛋白基因簇两类•类珠蛋白基因簇:16p122121基因有两个:1、2每个正常个体:212112/12•类珠蛋白基因簇:11p122GA14.Hb的发育演变与遗传控制2121GA1成人Hb珠蛋白链GowerⅠHb22基因2222222222GowerⅡPortlandFA2A转录翻译Hb类型胚胎Hb胎儿HbHb发育演变与遗传控制的特点⑴胚胎早期先合成和→HbGowerⅠ同时或稍后合成和→GowerⅡ、Portland⑵12周时和逐渐消失,链迅速增加,开始合成,HbF为主
⑶妊娠末期和出生不久,链迅速降低,链迅速增加,HbA为主
二.Hb病的分子遗传学Hb病的分类•异常Hb病:基因异常导致珠蛋白肽链结构和功能异常•地中海贫血:基因缺失或缺陷导致珠蛋白肽链合成量异常