药品生产质量管理规范附录一、总则1.本附录为国家药品监督管理局发布的《药品生产质量管理规范》(1998年修订)对无菌药品、非无菌药品、原料药、生物制品、放射性药品、中药制剂等生产和质量管理特殊要求的补充规定
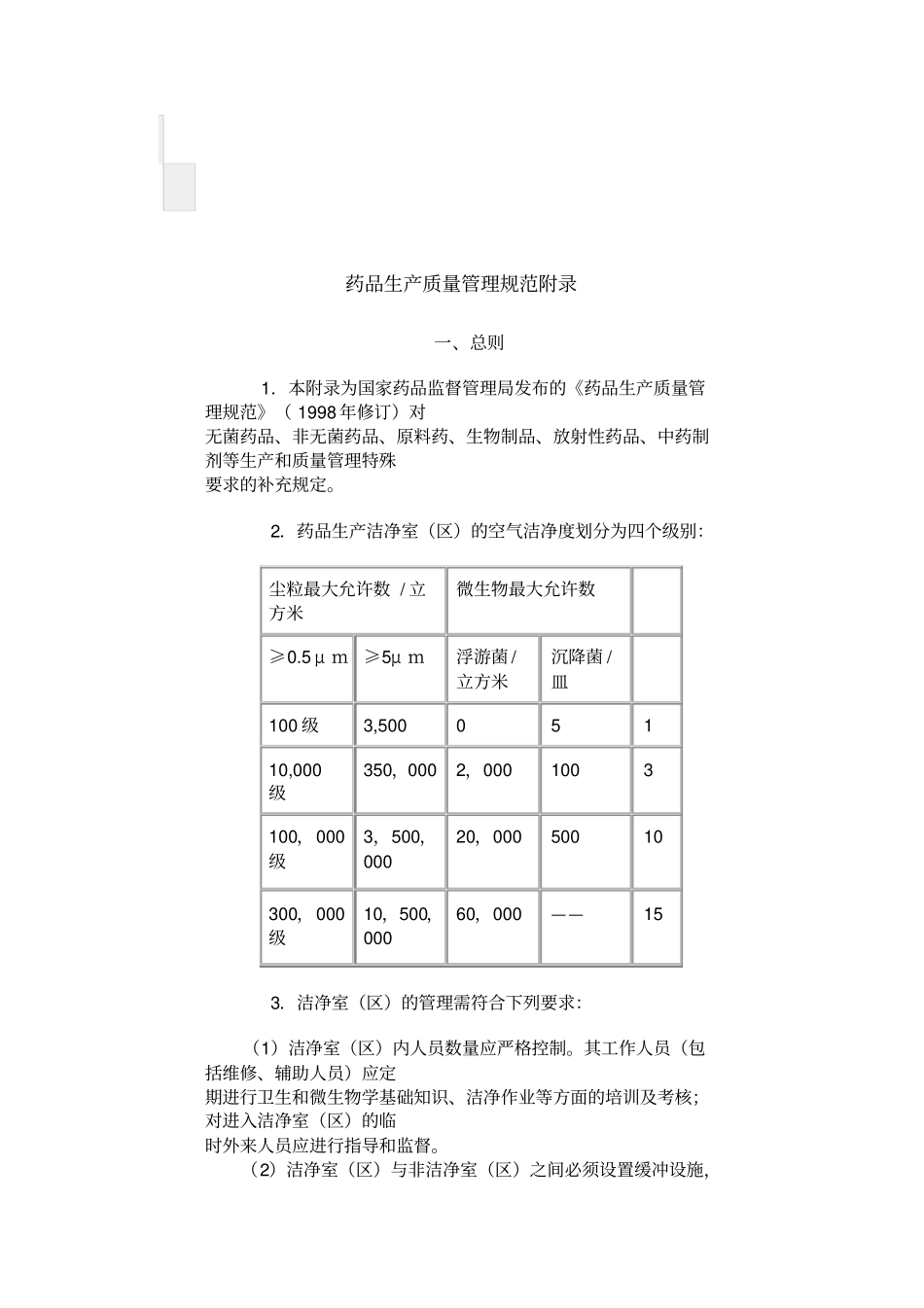
2.药品生产洁净室(区)的空气洁净度划分为四个级别:尘粒最大允许数/立方米微生物最大允许数≥0
5μm≥5μm浮游菌/立方米沉降菌/皿100级3,50005110,000级350,0002,0001003100,000级3,500,00020,00050010300,000级10,500,00060,000——153.洁净室(区)的管理需符合下列要求:(1)洁净室(区)内人员数量应严格控制
其工作人员(包括维修、辅助人员)应定期进行卫生和微生物学基础知识、洁净作业等方面的培训及考核;对进入洁净室(区)的临时外来人员应进行指导和监督
(2)洁净室(区)与非洁净室(区)之间必须设置缓冲设施,人、物流走向合理
(3)100级洁净室(区)内不得设置地漏,操作人员不应裸手操作,当不可避免时,手部应及时消毒
(4)10,000级洁净室(区)使用的传输设备不得穿越较低级别区域
(5)100,000级以上区域的洁净工作服应在洁净室(区)内洗涤、干燥、整理,必要时应按要求灭菌
(6)洁净室(区)内设备保温层表面应平整、光洁,不得有颗粒性物质脱落
(7)洁净室(区)内应使用无脱落物、易清洗、易消毒的卫生工具,卫生工具要存放于对产品不造成污染的指定地点,并应限定使用区域
(8)洁净室(区)在静态条件下检测的尘埃粒子数、浮游菌数或沉降菌数必须符合规定,应定期监控动态条件下的洁净状况
(9)洁净室(区)的净化空气如可循环使用,应采取有效措施避免污染和交叉污染
(10)空气净化系统应按规定清洁、维修、保养并作记录
4.药品生产过程的验证内容必须包括:(1)空气净化系统(2)工艺用水系统(3)生产工