Sybyl 对接教程1
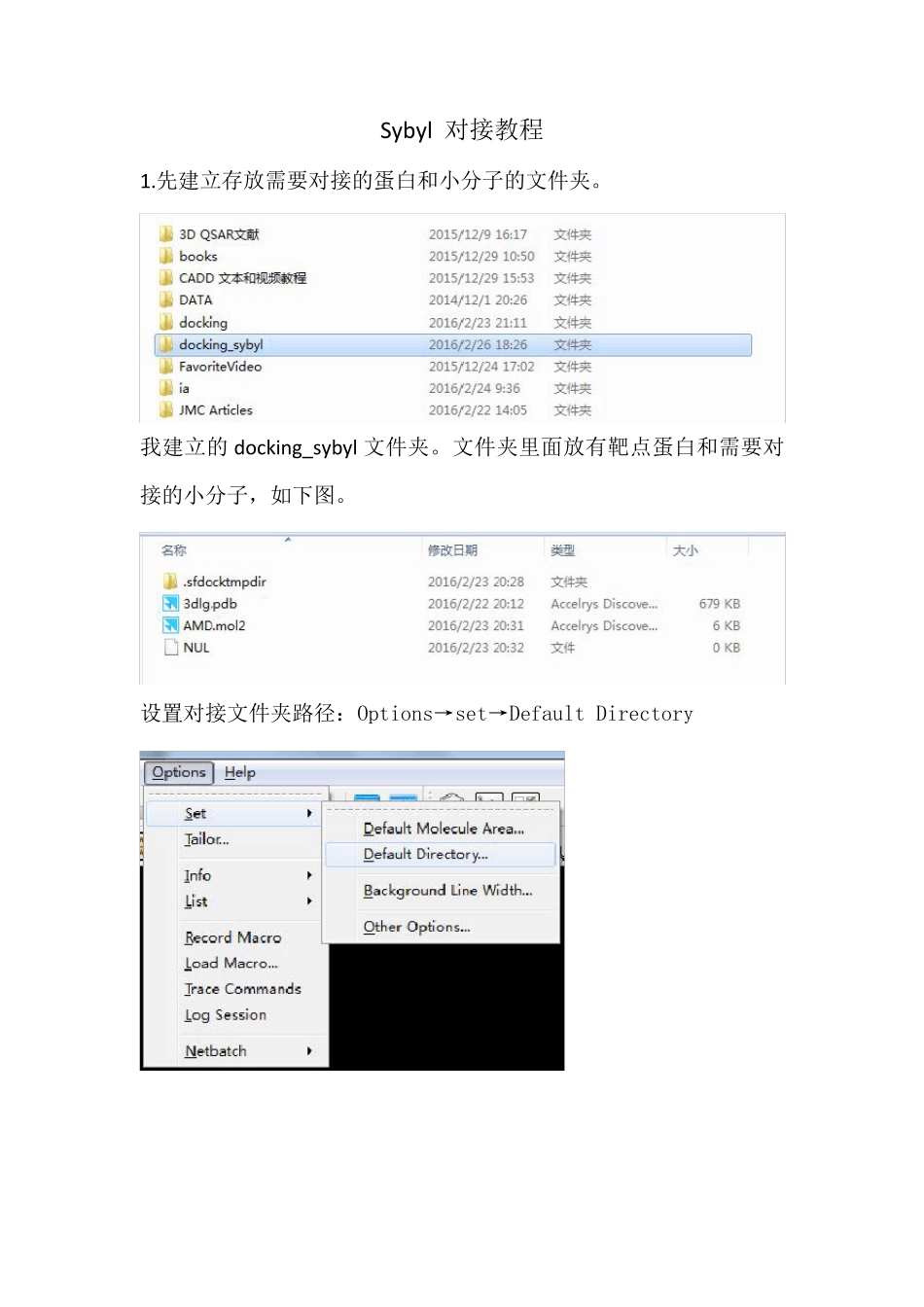
先建立存放需要对接的蛋白和小分子的文件夹
我建立的docking_sybyl 文件夹
文件夹里面放有靶点蛋白和需要对接的小分子,如下图
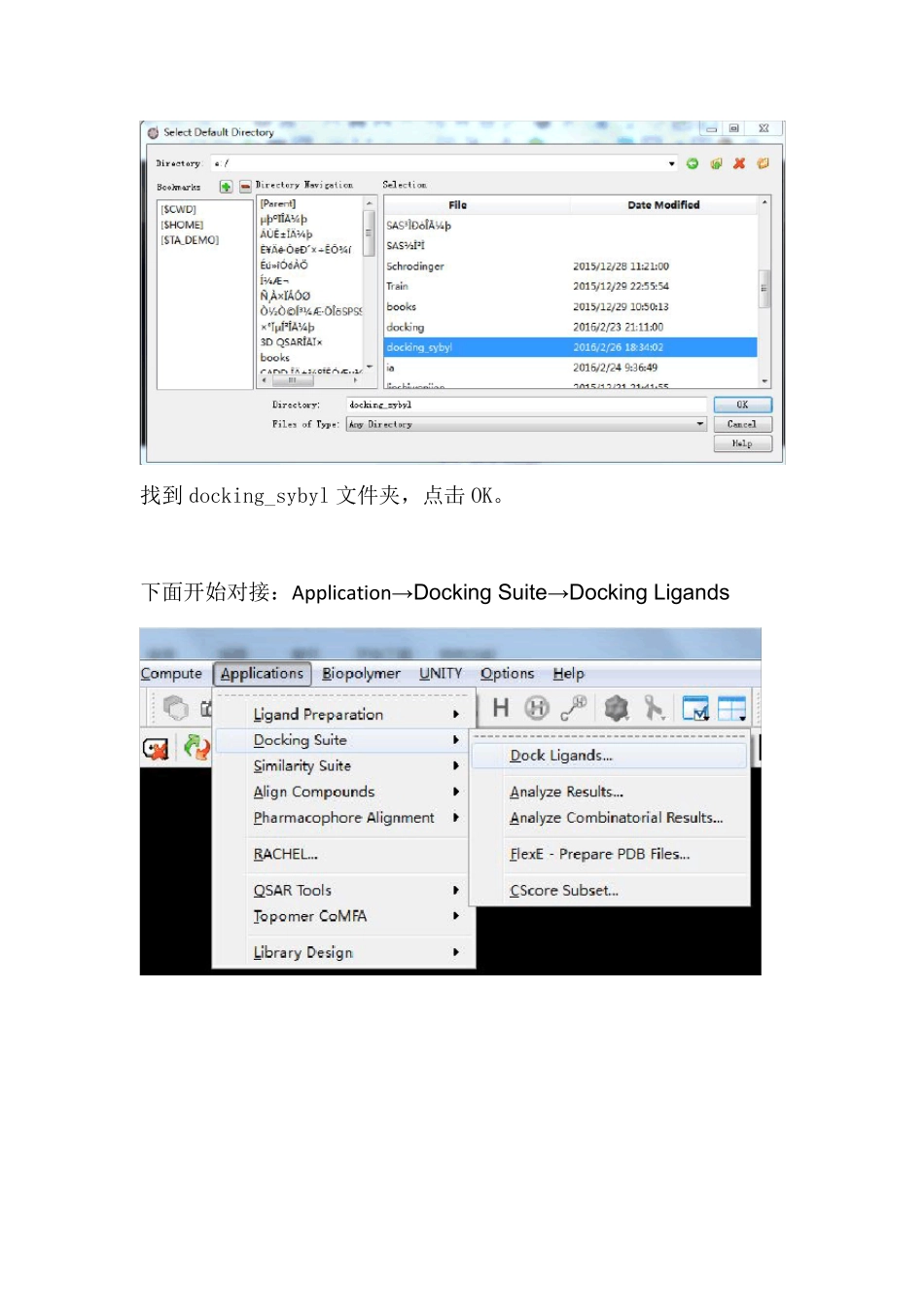
设置对接文件夹路径:Options→set→Default Directory找到docking_sybyl 文件夹,点击OK
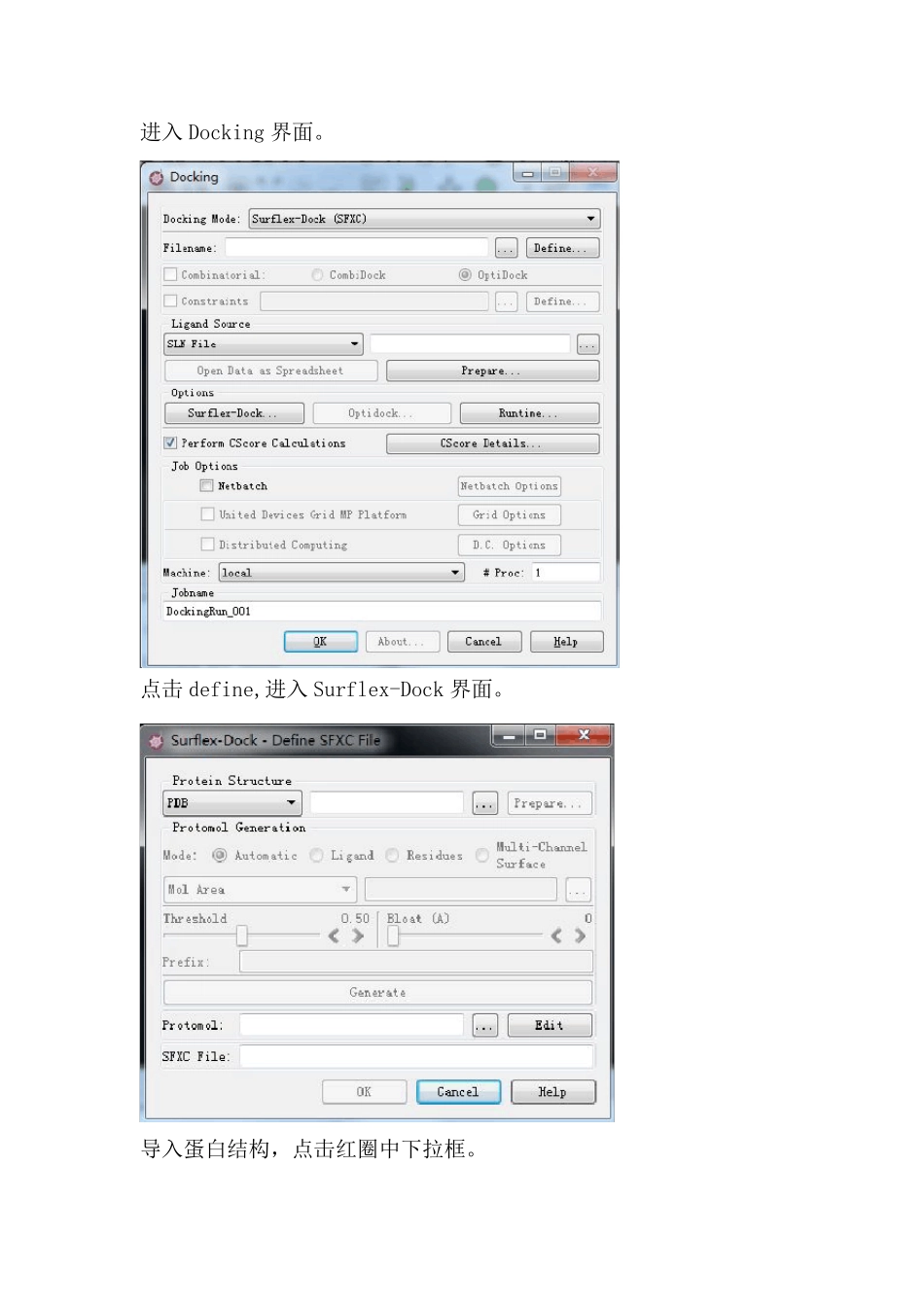
下面开始对接:Application→ Docking Suite→ Docking Ligands进入Docking 界面
点击 define,进入Surflex-Dock 界面
导入蛋白结构,点击红圈中下拉框
进入docking_sybyl 文件夹
选择 3dlg
pdb 点击 OK
这样蛋白已经导入sybyl 窗口
接下来对蛋白
点击 Prepare
点击Extract Ligand Substructures(提取小配体)
选择小配体GWE999,点击OK
点击Remove Substructures(删去无关的水分子和离子)点击OK到此蛋白已经准备好
点击Return
点击Generate,产生活性位点
即产生protomol,3dlg_H-L-0
50-0-protomol
mol2点击OK
在Ligand source 下拉框中有常见的小分子格式可以选择,看你要对接的小分子是什么格式就选什么格式
我的是mol2 格式就选择mol2 file点击红圈中下拉框,选择需要对接的小分子
mol2 点击OK
Proc 根据你的电脑的核数来确定,我的是两核,填2
Jobname 可以根据习惯自己命名
点击 OK,开始对接
对接完后出现Result Browser 界面
View 选择DockingRun1_site
mol2点击小分子AMD,出现结合模式
点击save result设置如下:每个配体保留3 个构象点击OK
显示小分子前3 个构象选择