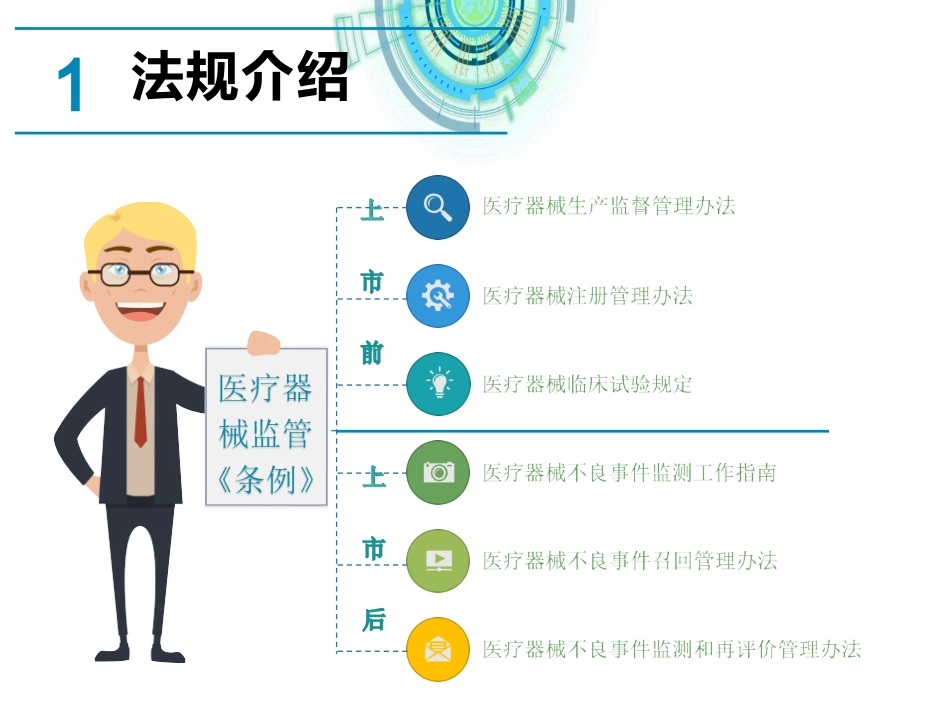
质管部xxx医疗器械不良事件目录法规介绍不良事件概念不良事件处理不良事件监测制度1法规介绍02概念(MDR)02概念(MDR)02概念(MDR)0103监管机构可以减少或避免同类医疗器械不良事件的发生,减低患者、医护人员使用医疗器械的风险,保障人民群众用械安全患者进一步提高医疗器械性能和功能的要求,推进企业对新产品的研制,有利于促进医疗器械行业的健康发展02企业03处置报告调查收集评价控制分析•医疗器械不良事件报告应当遵循可疑即报的原则报告原则•应当真实、完整、准确,导致死亡、严重伤害或者可能导致严重伤害或者死亡的医疗器械不良事件应当报告报告内容•在首个注册周期内,应当报告所有医疗器械不良事件创新医疗器械ABCDEF导致死亡应当在7日内报告导致严重伤害、可能导致严重伤害或者死亡20日内报告产品在境外发生导致或者可能导致严重伤害或者死亡境外注册人指定的代理人和国产医疗器械注册人应当自发现或者获知之日起30日内报告32145630日内45日内导致严重伤害、可能导致严重伤害或者死亡的事件导致死亡的事件基础深入分析讨论措施报告调查核实监测数据、产品检验情况和其他风险专家讨论会(必要时)风险控制措施通过系统提交主动召回医疗器械生产企业按照本办法第十条、第十二条的要求进行调查评估后,确定医疗器械产品存在缺陷的,应当立即决定并实施召回,同时向社会发布产品召回信息
食品药品监督管理部门经过调查评估,认为医疗器械生产企业应当召回存在缺陷的医疗器械产品而未主动召回的,应当责令医疗器械生产企业召回医疗器械
责令召回C注册人应当及时对监测哨点收集到的死亡、严重伤害或者可能导致严重伤害或者死亡的不良事件进行调查
DE注册人应当对监测哨点收集的不良事件进行汇总,结合系统反馈数据和文献资料进行风险分析,评估不良事件风险特征、严重程度、不良事件发生率等风险状况注册人应当从重点监测品种的结构设计、原材料成分