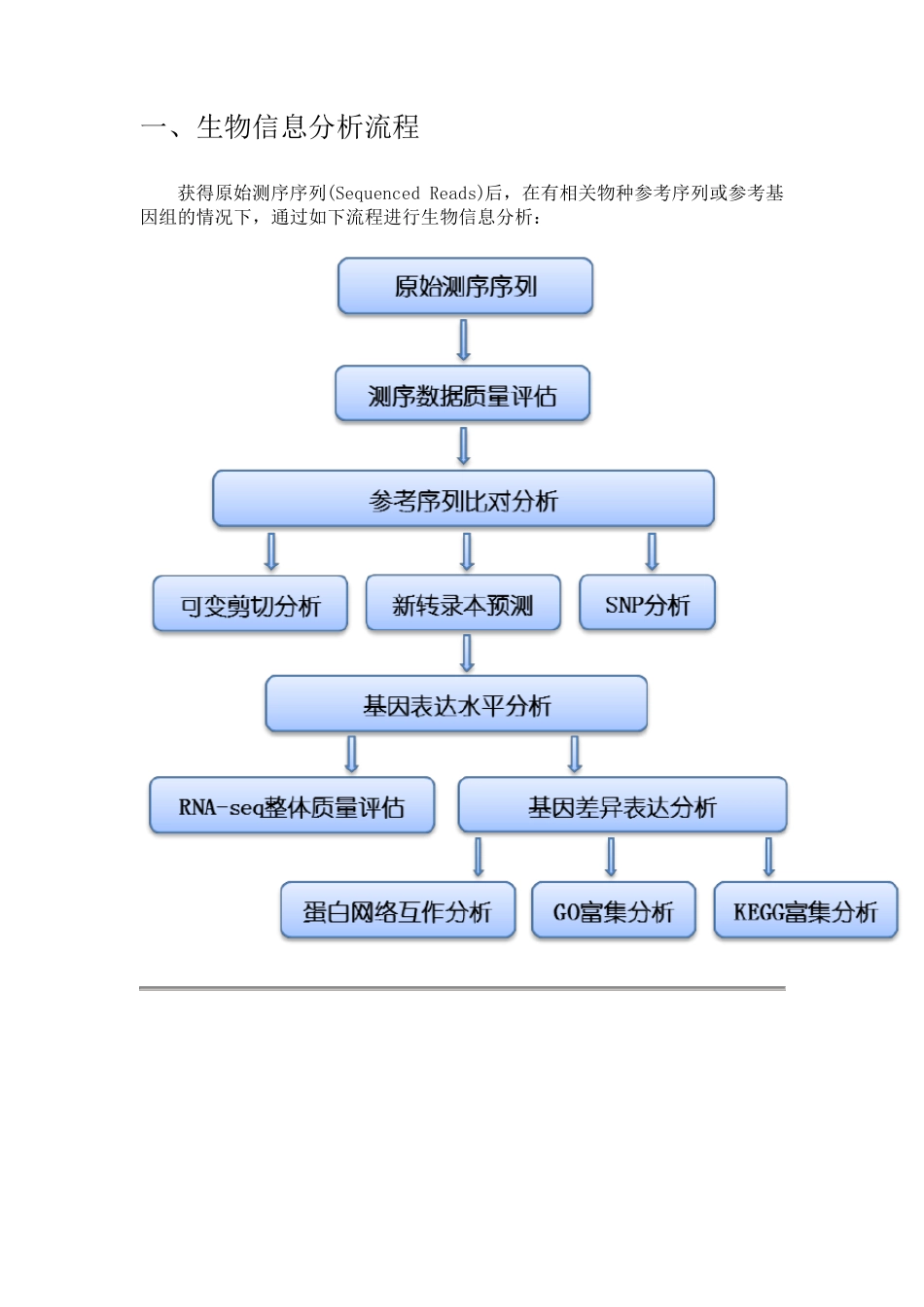
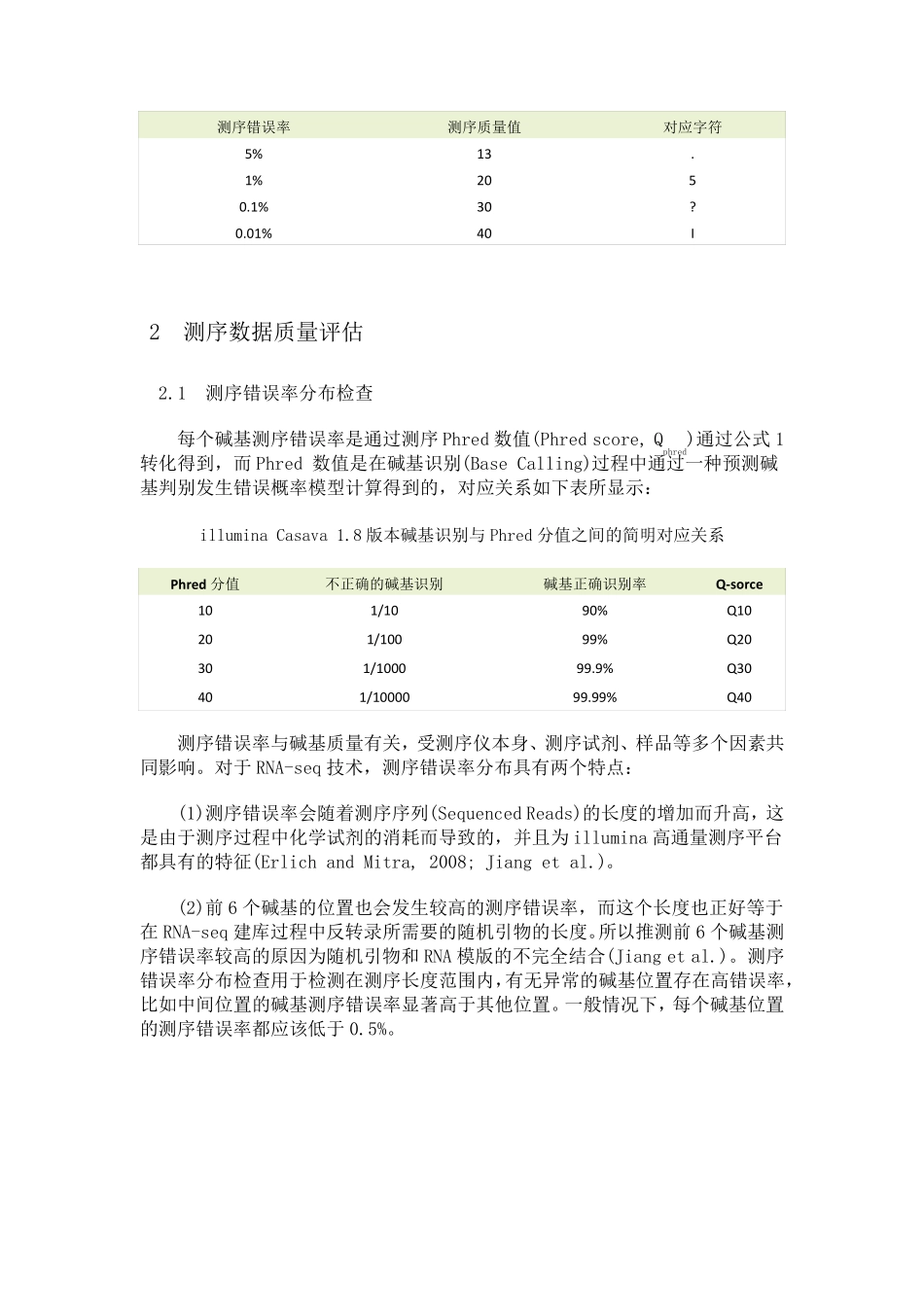
一、生物信息分析流程 获得原始测序序列(Sequenced Reads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析: 二、项目结果说明 1 原始序列数据 高通量测序(如illumina HiSeqTM2000/MiSeq 等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data 或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息
FASTQ 格式文件中每个read 由四行描述,如下: @EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG GCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT + @@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF 其中第一行以“@”开头,随后为illumina 测序标识符 (Sequence Identifiers) 和描述文字 (选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al
illumina 测序标识符详细信息如下: EAS139 Unique instrument name 136 Run ID FC706VJ Flowcell ID 2 Flowcell lane 2104 Tile number within the flowcell lane 15343 'x'-coordinate of the cluster within the