同源模建的方法与结果分析Version1
2-------------------------------------------------------序言:作为一个以实验为主的生化工作者来说,很多时候可以通过分子生物学手段获取自己需要的目的基因,并在各种表达载体和宿主中进行对应蛋白的表达,随后对于这些蛋白的特性进行研究,这也是一般酶学研究的特定套路
而近十几年来,人们开始思考是否能够将特性与蛋白质的三级结构进行关联,从分子水平理解蛋白质与底物之间的相互作用呢于是类似于蛋白结构模建、分子对接、分子动力学模拟、量化计算等多种手段相继被创造以及应用
在这些方法中,同源模建无疑是最基础也是最重要的一个步骤,因为其质量的好坏直接决定了后续工作是否可信
因此,本文打算就同源模建的基本原理、常用软件及服务器以及结果分析与改进提供一些个人的经验,并希望各位朋友能够给予批评指正
同源模建的原理及应用限制两点基本原理:1
一个蛋白质的结构由其氨基酸序列唯一的决定
知道其一级序列,至少在理论上足以获取其结构2
结构在进化中更稳定,变化比序列层面的变化要缓慢许多
应用限制:模板蛋白和目标蛋白的序列一致性需要大于30%,且越大建模准确性越有保障
了解了基本的原理,我们需要知道在实际操作中,同源模建都需要怎么样进行
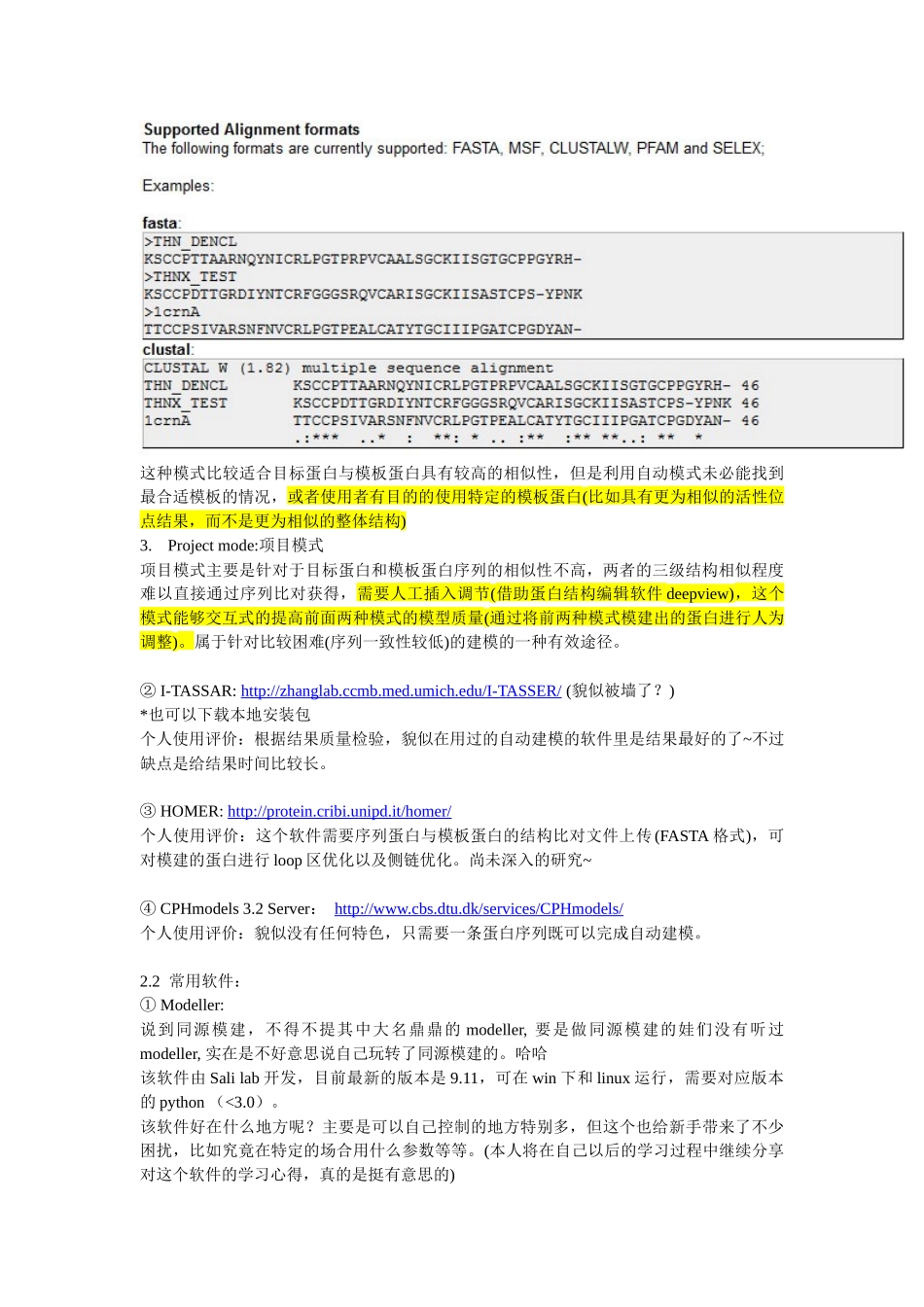
同源模建的过程从实践中可分为以下7个步骤:1.模板识别和初始比对在序列一致性比较高的时候,可以通过简单的序列比对程序如BLAST获取目标蛋白的结构(将比对的数据库选择为PDB数据库)
2.比对结果的校正用以上的方法确定一个或多个建模模板后,应该采用更为精确的方法已取得更优的比对结果
有时在序列一致性较低的区域比对两条序列可能会具有困难,这个时候,我们可以采取其他同源蛋白序列一起参与比对来找到解决的办法
3.主链生成比对完成后,就可以开始实际的建模过程了,相对与后面几步来说,主链