GMP 对制药厂房设计的要求 a(67 页)Good is good, but better carries it
精益求精,善益求善
GMP 对制药厂房设计的要求1
总则制药厂房的工程设计是为药品生产能达到保障质量要求而制造合格的布局、合理的生产场所
它是指制剂、原料药、药用辅料和直接接触药品的包装材料生产中所需的建筑物以及与工艺配套的空气调节、水处理等公用工程
GMP 要求制药企业消除混药和污染,最大限度地减少任何药品生产所包含的、通过检验最终产品不能消除的风险
要做到符合生产流程要求并防止交叉污染和混杂(所谓混杂是指因厂房平面布局不当及管理不严,造成不合格的原料、中间体及半成品的误操作使不合格品继续加工包装出厂,或生产中遗漏任何生产程序或控制步骤),厂房的合理布局显得特别重要,在进行新厂建设或老厂改造时,必需仔细地反复推敲平面布置
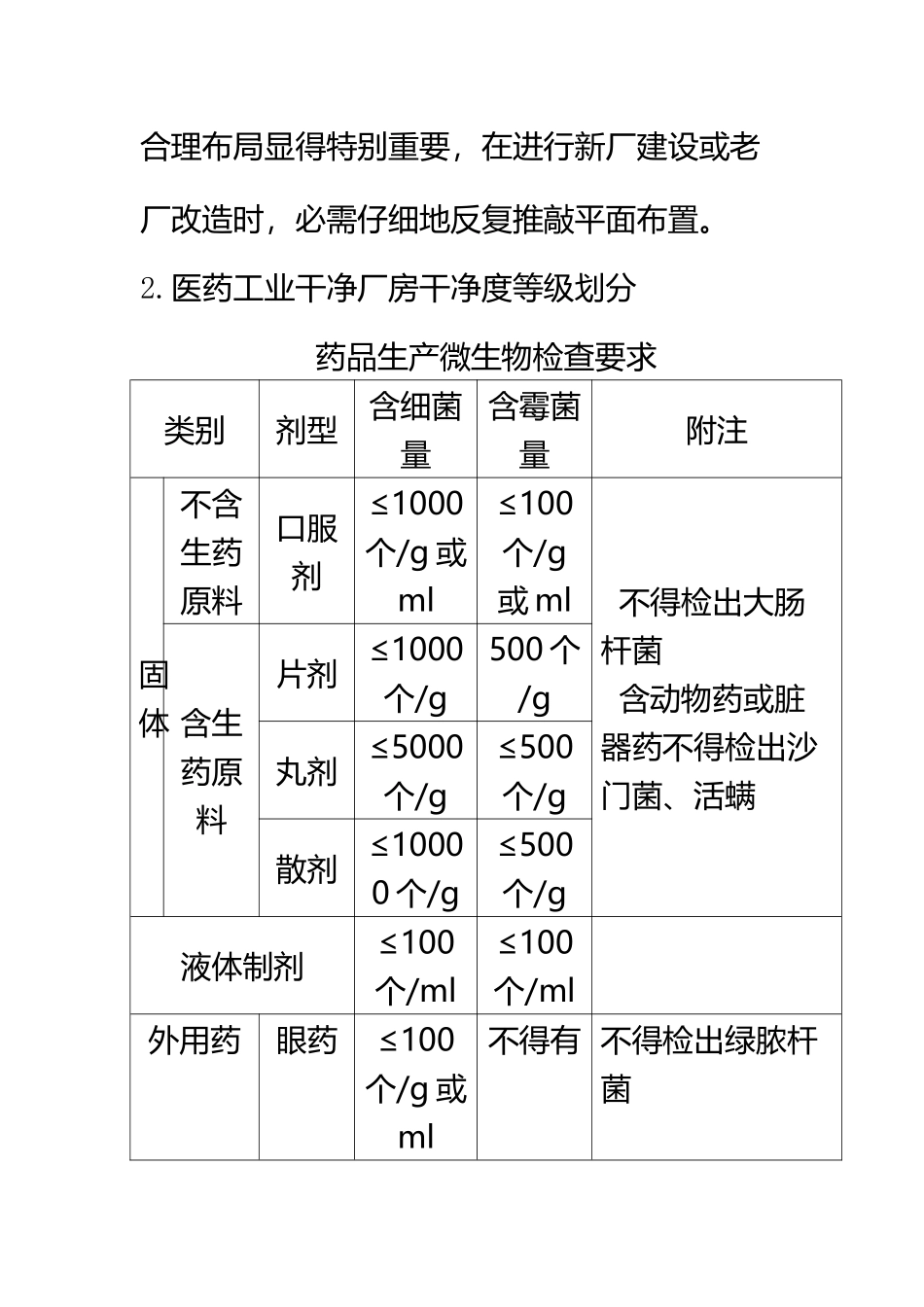
医药工业干净厂房干净度等级划分药品生产微生物检查要求类别剂型含细菌量含霉菌量附注固体不含生药原料口服剂≤1000个/g 或ml≤100个/g或 ml 不得检出大肠杆菌 含动物药或脏器药不得检出沙门菌、活螨含生药原料片剂≤1000个/g500 个/g丸剂≤5000个/g≤500个/g散剂≤10000 个/g≤500个/g液体制剂≤100个/ml≤100个/ml 外用药眼药≤100个/g 或ml不得有 不得检出绿脓杆菌阴道创伤用药≤1000个/g 或ml≤100个/g或 ml不得检出破伤风杆菌、绿脓杆菌
药品生产干净室(区)空气干净度级别干净度级别尘粒最大允许数/个
m-3微生物最大允许数≤0
5μm≥5μm浮游菌/个
m-3沉降菌/个
皿-1100 级350005110 000级3500020001003100 000 级35000002000050010300 000 级1050000060000 15注:① 300 000 级是参考美