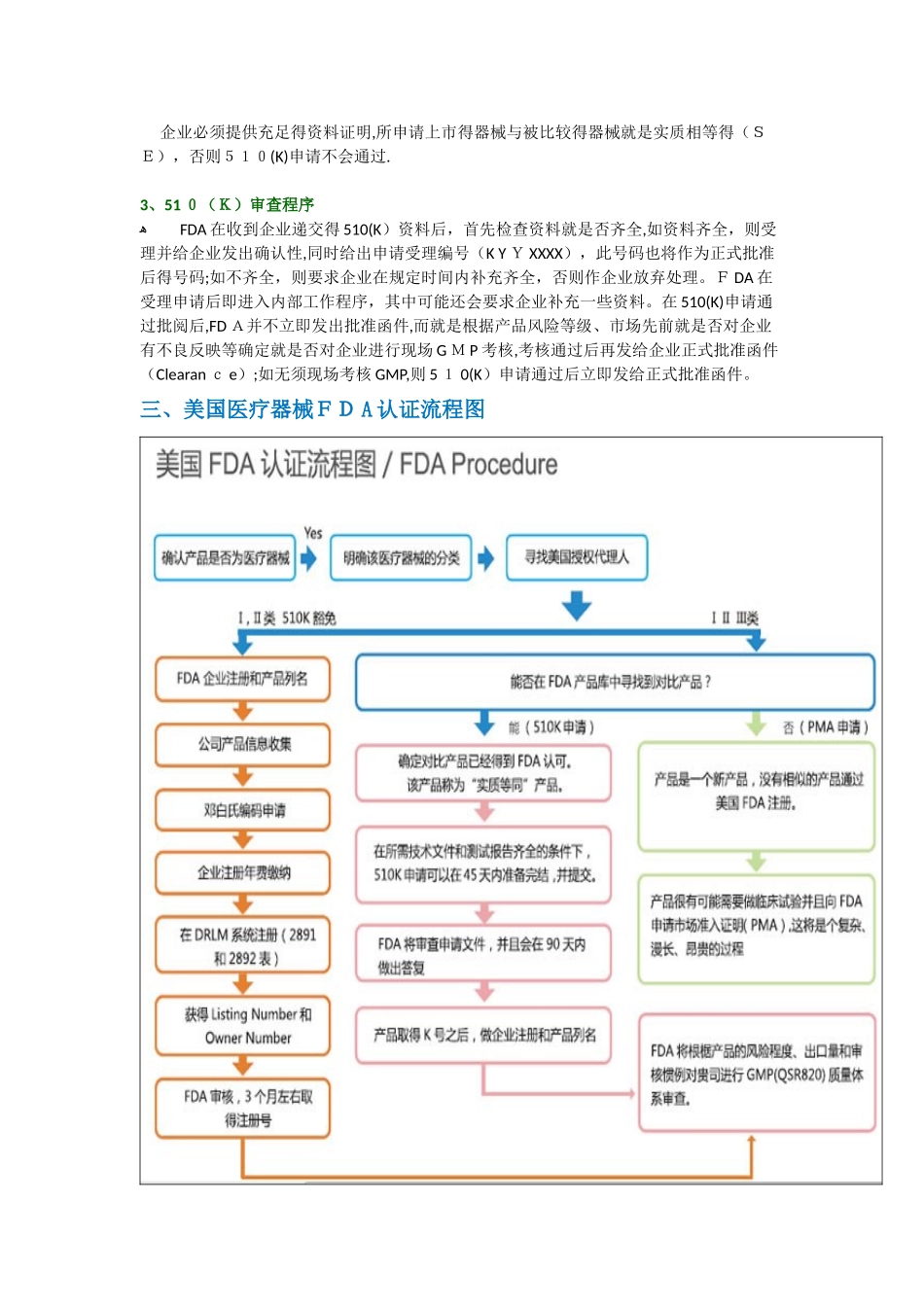
美国医疗器械 F D A 认证流程一、美国医疗器械 F D A 认证介绍 FDA对医疗器械得管理通过器械与放射健康中心进行得,中心监督医疗器械得生产、包装、经销商遵守法律下进行经营活动
医疗器械范围很广,小到医用手套,大至心脏起博器,均在FDA监督之下,根据医疗用途与对人体可能得损害,FDA将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,越高类别监督越多、 假如产品就是市场上不曾存在得新颖发明,FDA要求厂家进行严格得人体实验,并有令人信服得医学与统计学证据说明产品得有效性与安全性
医疗器械得FDA认证,包括:厂家在FDA注册、产品得FDA登记、产品上市登记(510表登记)、产品上市审核批准(PMA审核) 医疗保健器械得标签与技术改造、通关、登记、上市前报告,须提交以下材料:(1)包装完整得产成品五份,(2)器械构造图及其文字说明,(3)器械得性能及工作原理;(4)器械得安全性论证或试验材料,(5)制造工艺简介,(6)临床试验总结,(7)产品说明书、 如该器械具有放射性能或释放放射性物质,必须详细描述、 医疗器械得工厂与产品注册FDA对医疗器械有明确与严格得定义,其定义如下:“所谓医疗器械就是指符合以下条件之仪器、装置、工具、机械、器具、插入管、体外试剂及其它相关物品,包括组件、零件或附件:明确列于National Formulary或the Unite States Pharmacopeia或前述两者得附录中者;预期使用于动物或人类疾病,或其它身体状况之诊断,或用于疾病之治愈、减缓与治疗者;预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目得者”
只有符合以上定义得产品方被瞧作医疗器械,在此定义下,不仅医院内各种仪器与工具,即使连消费者可在一般商店购买之眼镜框、眼镜片、牙刷与按摩器等健身器材等都属于FDA之管理范围
它与国内对医疗器械得认定稍有不同
根据风险等级得不同,F