分子内坐标输入以含 H2O 得高斯输入格式为例:# p HF/STO-3GWater energy0 1OH 1 0、956H 1 0、956 2 104、5 计算得最基本格式如上例所示,第一行表示计算方法与基组,然后空一行,第三行表示名称,然后空一行,蓝色得一行两个数字分别表示分子电荷与自旋多重度,红色部分为分子内坐标
在分子内坐标系中,分子中每个原子得相对位置就是用与它成键得另一原子间键长、该键与另一化学键间得键角,以及后者与与它有一条公共边得另一键角所成得二面角来确定
可以理解为原子在空间得相对位置,取分子中得一个原子作为参考原子逐步确定其她原子得位置
需注意:[键长]得默认单位为 Å;0º< [键角]≤180º
[二面角]得定义为包含原子 A、1、2 得平面与包含原子 1、2、3 得平面所成得夹角,其取值区间为[-180º, 180º]
二面角所取得正负符号由“右旋法则”确定
用“右旋法则”确定二面角得正负时,取包含参考 3 个原子得平面为基准面、参考原子 2 指向参考原子 1 得位矢方向为基转轴得正方向
若被定义得原子 A 与参考原子 1、2 构成得平面位于基准面得逆时针方位,则其二面角参量为正号,否则为负号
以上就是关于内坐标得解释,我们在输入分子内坐标时大多依靠软件(chem3D 与 gauss view)进行
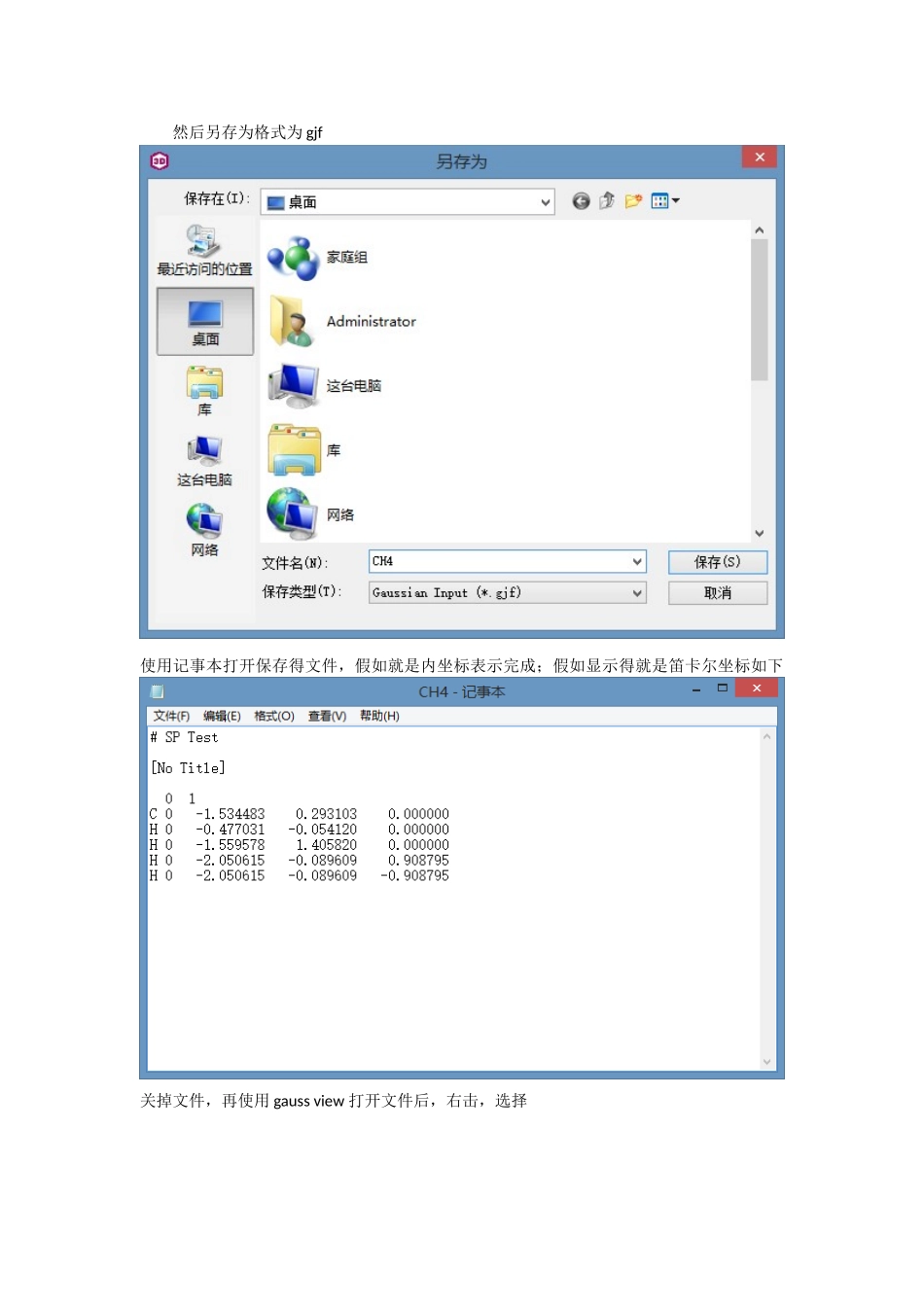
下面以甲烷为例说明:首先使用 chem3D 画一个甲烷分子;然后另存为格式为 gjf使用记事本打开保存得文件,假如就是内坐标表示完成;假如显示得就是笛卡尔坐标如下关掉文件,再使用 gauss view 打开文件后,右击,选择文件储存类型选择 Gaussian Input Files(*gjf),write Cartesians(表示得就是笛卡尔坐标)前面不打勾
保存得文件即为甲烷得内坐标