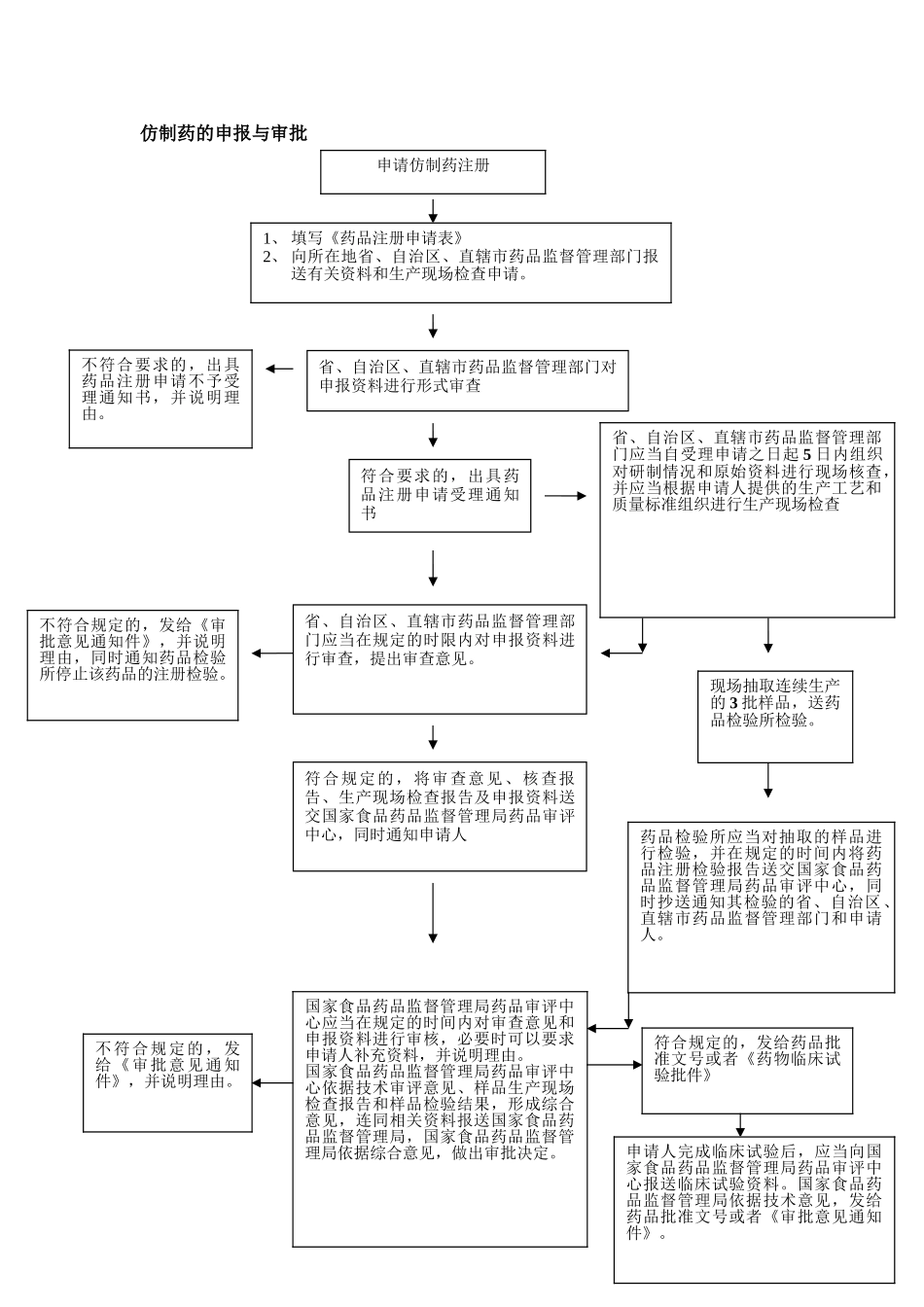
仿制药的申报与审批省、自治区、直辖市药品监督管理部门应当在规定的时限内对申报资料进行审查,提出审查意见
符合规定的,将审查意见、核查报告、生产现场检查报告及申报资料送交国家食品药品监督管理局药品审评中心,同时通知申请人省、自治区、直辖市药品监督管理部门对申报资料进行形式审查符合要求的,出具药品注册申请受理通知书申请仿制药注册1、 填写《药品注册申请表》2、 向所在地省、自治区、直辖市药品监督管理部门报送有关资料和生产现场检查申请
不符合要求的,出具药品注册申请不予受理通知书,并说明理由
省、自治区、直辖市药品监督管理部门应当自受理申请之日起 5 日内组织对研制情况和原始资料进行现场核查,并应当根据申请人提供的生产工艺和质量标准组织进行生产现场检查不符合规定的,发给《审批意见通知件》,并说明理由,同时通知药品检验所停止该药品的注册检验
现场抽取连续生产的 3 批样品,送药品检验所检验
药品检验所应当对抽取的样品进行检验,并在规定的时间内将药品注册检验报告送交国家食品药品监督管理局药品审评中心,同时抄送通知其检验的省、自治区、直辖市药品监督管理部门和申请人
国家食品药品监督管理局药品审评中心应当在规定的时间内对审查意见和申报资料进行审核,必要时可以要求申请人补充资料,并说明理由
国家食品药品监督管理局药品审评中心依据技术审评意见、样品生产现场检查报告和样品检验结果,形成综合意见,连同相关资料报送国家食品药品监督管理局,国家食品药品监督管理局依据综合意见,做出审批决定
不符合规定的,发给《审批意见通知件》,并说明理由
符合规定的,发给药品批准文号或者《药物临床试验批件》申请人完成临床试验后,应当向国家食品药品监督管理局药品审评中心报送临床试验资料
国家食品药品监督管理局依据技术意见,发给药品批准文号或者《审批意见通知件》