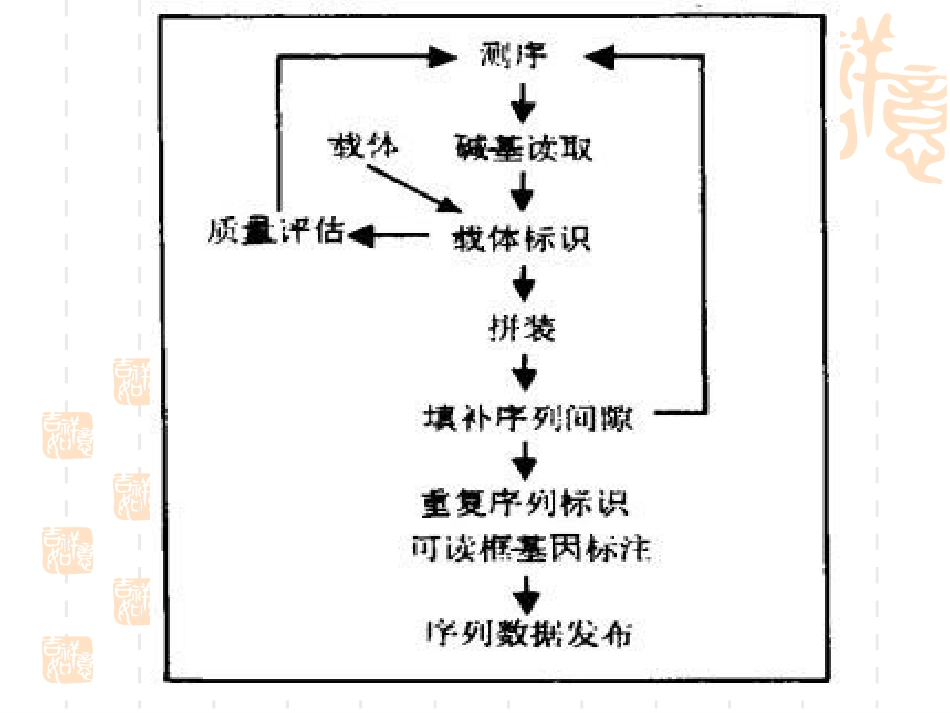
基因组序列拼接段倩倩序列拼接序列拼接任务即将测序生成的reads短片段拼接起来,恢复出原始的序列
该问题是序列分析的最基本任务,是基因组研究成功与失败的关键,拼接结果直接影响到序列标注,基因预测、基因组比较等后续任务
基因组序列的拼接也是基因组研究必须解决的首要难题
其困难不仅来自它的海量数据(以人类基因组序列为例,从数量为10兆级的片断恢复出长度为亿级的原始序列),而且源于它含有高度重复的序列
拼接问题的难点DNA测序数据有其固有的四个的特点,他们也正是解决实际的序列拼接问题的难点所在:1.测序有误差2.不完全覆盖性3.序列所在链不确定4.重复序列的干扰1.测序有误差由于测序技术的局限,难免会出现测序错误,尤其是在序列的末端,一般错误率可控制在1%以下
所以对每个碱基一般有一个正确概率,以质量打分的形式给出
因此每个ri都有个可信度
而read与read之间有不同程度的重叠,由此导致有的重叠可信度高,有的重叠可信度低
2.不完全覆盖性不是所有的碱基被测序的次数都等于平均测序覆盖度
极端的情况,可能会出现源基因组序列上部分区域未被测序的情况(这段区域称为gap)
即,测序的reads集合不是原始基因组序列一个完整覆盖
此时需要借助于各种图谱如:基因组指纹图谱(genomefingerprintmap),基因组级物理图谱(genome-widephysicalmap),细胞发生图谱(cytogeneticmaps)等协助对reads进行定位.3.序列所在链不确定由于测序过程中无法确定特定片断属于DNA双链中的哪一条链上,所以我们在拼接过程中并不清楚使用的是read的正义链,还是其互补链
4.重复序列的干扰DNA序列自身含有高度重复的子序列,它们一种表现为短序列的串级重复,比如:(GGAA)n
或AmTn等
另一种表现为大量相似序列(其拷贝数可达几十万)散布在基因组的各个地方