如何计算HOMO、LUMO-ADF 1 ,导入分子 此处假定分子已经是优化好的结构,如果结构没有优化好,那么可以参考ADF 结构优化
把要计算的分子的坐标,从文本文件中复制: 打开 ADFinput:(windows:在开始菜单中找到 ADFinput 图标并点击,即打开 ADF 的图形界面;Linux:如果在 Linux服务器的桌面,那么在命令行输入ADFinput 即打开图形界面)
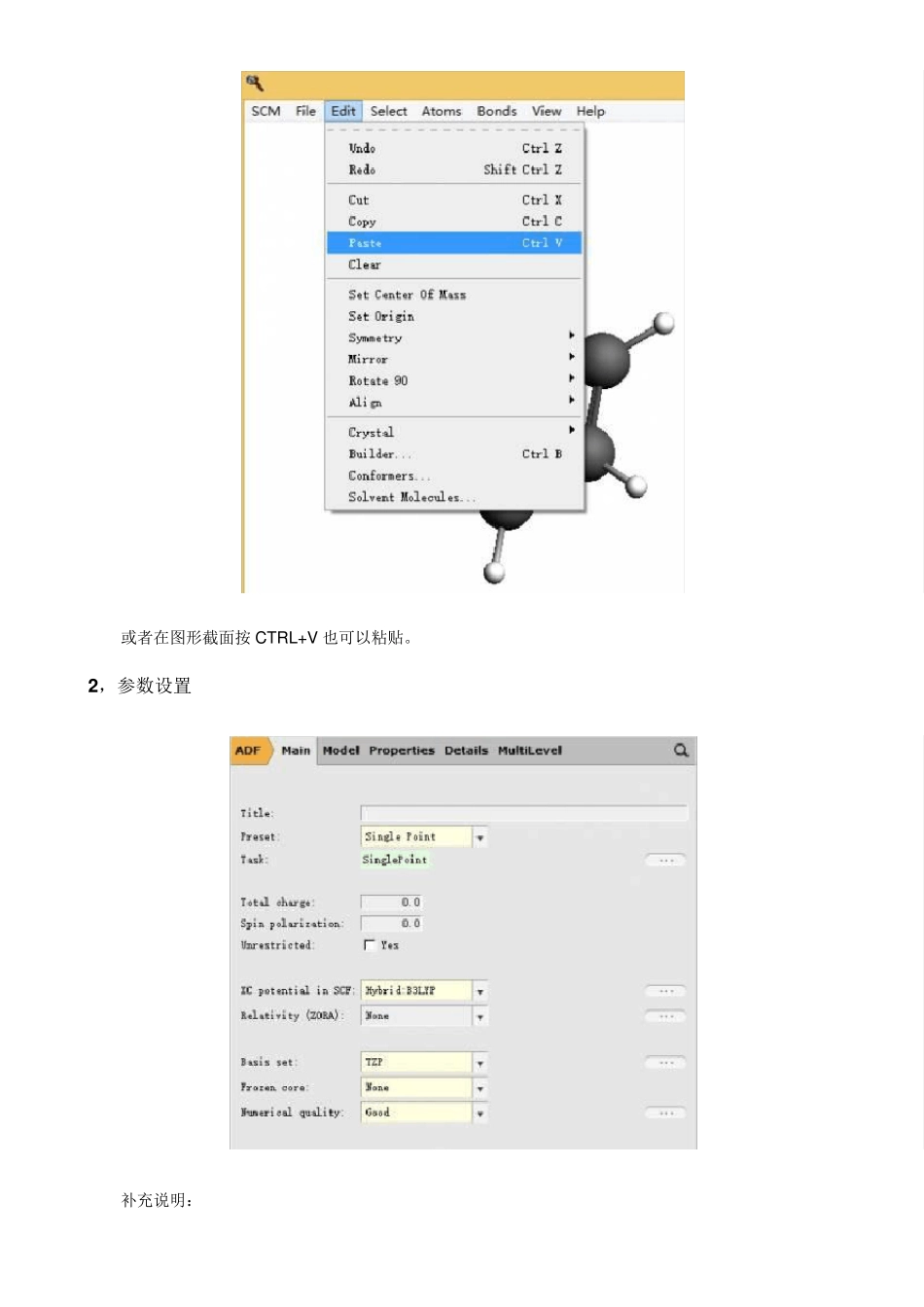
粘贴到 ADF 图形界面中: 或者在图形截面按CTRL+V 也可以粘贴
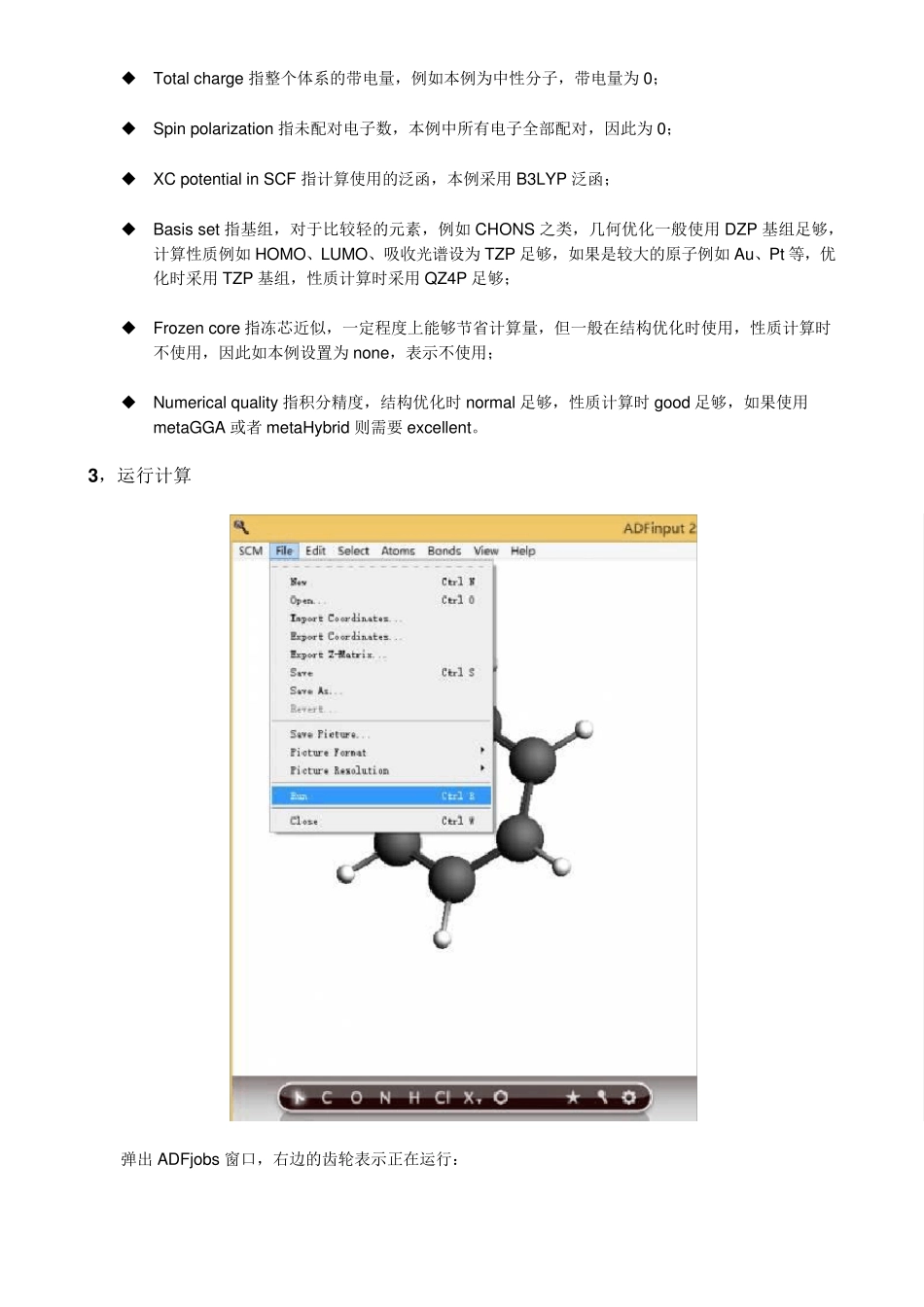
2 ,参数设置 补充说明: Total charge 指整个体系的带电量,例如本例为中性分子,带电量为0; Spin polarization 指未配对电子数,本例中所有电子全部配对,因此为0; XC potential in SCF 指计算使用的泛函,本例采用B3LYP 泛函; Basis set 指基组,对于比较轻的元素,例如CHONS 之类,几何优化一般使用DZP 基组足够,计算性质例如HOMO、LUMO、吸收光谱设为TZP 足够,如果是较大的原子例如Au、Pt 等,优化时采用TZP 基组,性质计算时采用QZ4P 足够; Frozen core 指冻芯近似,一定程度上能够节省计算量,但一般在结构优化时使用,性质计算时不使用,因此如本例设置为none,表示不使用; Numerical quality 指积分精度,结构优化时 normal 足够,性质计算时 good 足够,如果使用metaGGA 或者 metaHybrid 则需要 excellent
3 ,运行计算 弹出 ADFjobs 窗口,右边的齿轮表示正在运行: 运行完毕后,变成实心球: 4,查看HOMO、LUMO 第二列的能级是分子的能级,第一列,以及后面所有列,都是原子的能级,这个图反应分子轨道与原子轨道的关系
鼠标放到感兴趣的