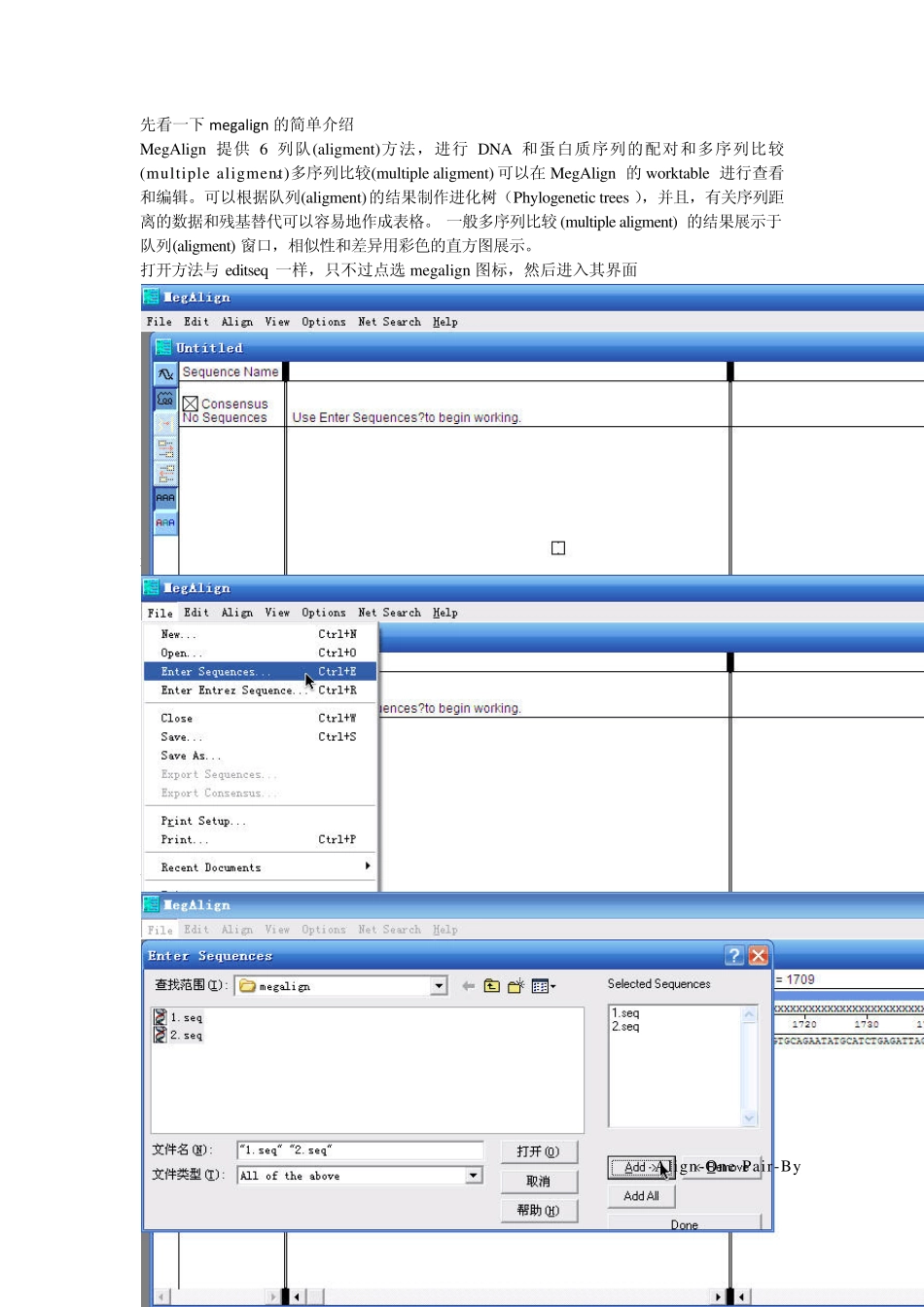
先看一下megalign的简单介绍 MegAlign 提供6 列队(aligment)方法,进行DNA 和蛋白质序列的配对和多序列比较(multiple aligment)
多序列比较(multiple aligment) 可以在 MegAlign 的worktable 进行查看和编辑
可以根据队列(aligment)的结果制作进化树(Phylogenetic trees ),并且,有关序列距离的数据和残基替代可以容易地作成表格
一般多序列比较(multiple aligment) 的结果展示于队列(aligment) 窗口,相似性和差异用彩色的直方图展示
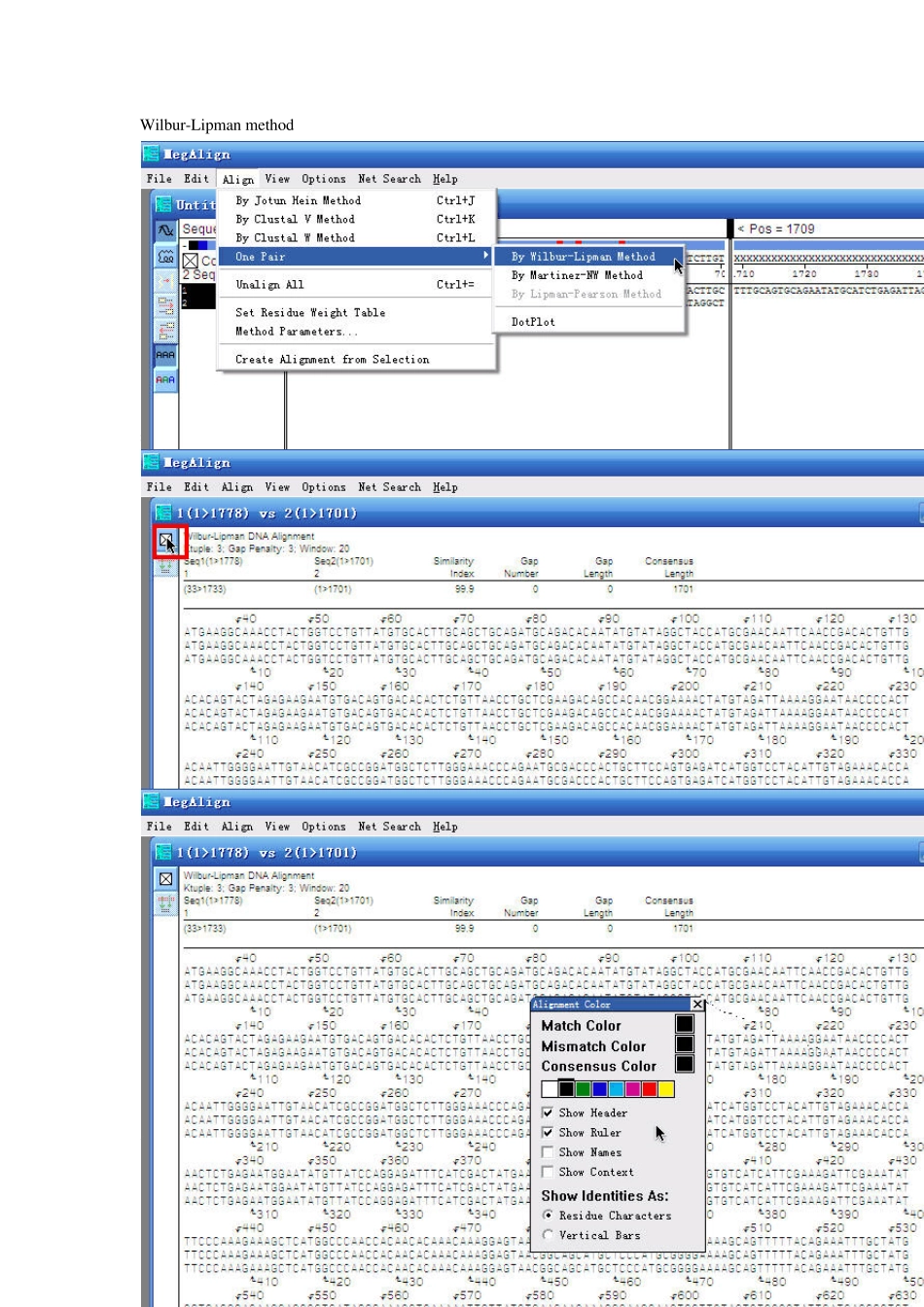
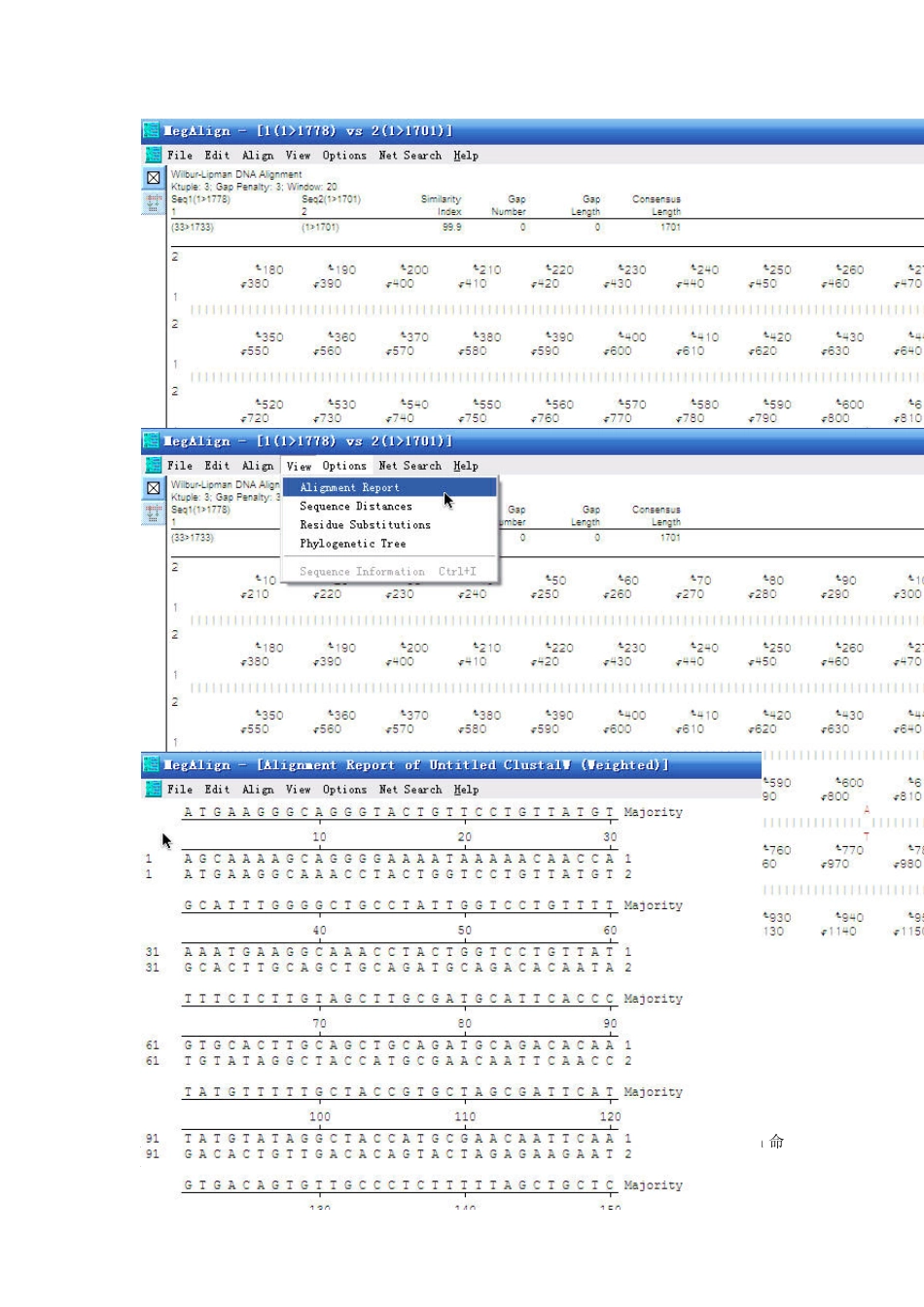
打开方法与 editseq 一样,只不过点选 megalign 图标,然后进入其界面 选择 File-Enter Sequences 首先进行2 个序列比对,选中所需序列1 和2,点击 add,使从左侧添加到右边的框中,单击 Done 出现如图所示界面,选中 1 与 2(可按 control 点选),之后选择 Align-One Pair-By Wilbu r-Lipman method 出现如图所示界面,即为 blast 结果,但画面不美观,可对其进行调整,点击鼠标所处位置按钮 出现此对话框,里面可进行一系列设置,可根据自己喜好进行,使界面更美观形象 设置后可看到错配碱基,如下,还是比较直观吧 比对之后可对其进行结果查看,点选 View -Alignment report 即可 结果如图 对于多序列的比对,添加序列与一对序列一样,不过选择的 Align-Clustal 或者 Jotun Hein 命令 如点了之后出现点选Jotun He2 次变成4 条后点选View-P现如下结果,ein 后,出现如条了 ) Phylogenetic因我用序列如图界面,图 Tree 进行系列太少,体现图中红线部分系统树分析 不出很