13 临床试验设计 中山大学公共卫生学院 医学统计学与流行病学系 骆福添 · 特点: (1)伦理:病人知情、同意、得益、退出自由 (2)法规:资格证书,设计格式化、实施不走样、资料样式化、统计规范化 (3)数据:合格集、意向集(+脱落)、安全集(+脱落+剔除) (4)规范:统计学规范—资料可信、基线可比、临床有效、使用安全 例:某制药企业从植物中提取了一个新的分子单体,品名“A 脂” ,药理学研 究证实了其降脂作用,主要能够降低低密度脂蛋白(LDLC)水平,毒理 学研究发现该药有轻微胃肠道不良反应,主要表现为腹胀和腹泻
企业 希望进行临床试验,把“A 脂” 开发成能够上市的新药
1 临床试验前的必要准备 13
1 临床试验必须符合法规要求 新药临床试验在开始前,必须得到药品监督管理部门的批准,批准的标 志是获得“新药临床研究批件”
临床试验的组织实施,也必须符合相关 的法规要求
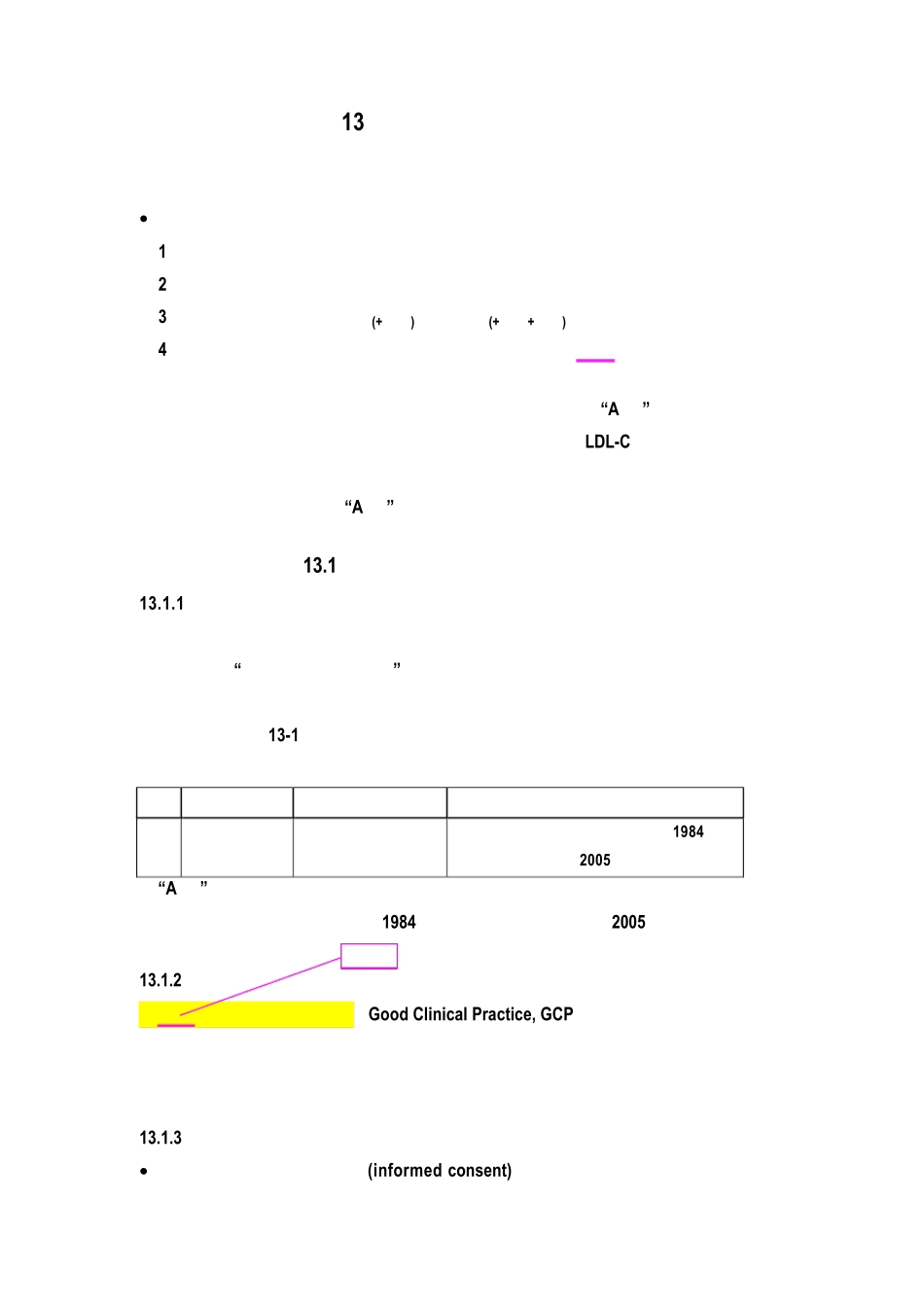
表 131 中国、美国、欧盟和日本的管理当局、 具体管理部门和主要相关法规 地区 管理当局 管理部门 主要法规 中国 国家食品药品 监督管理局 国家食品药品监督管 理局注册司 《中华人民共和国药品管理法》 1984、《药 品注册管理办法》2005 “A 脂” 如果在中国注册, 其临床试验必须符合中国相关法规的要求, 如 《中 华人民共和国药品管理法》1984、《药品注册管理办法》2005 等
2 临床试验需要符合相关指导原则要求 《药品临床试验管理规范》 (Good Clinical Practice, GCP)是临床试验的重要 指导原则
《临床试验统计学指导原则》 13
3 临床试验必须符合道德规范 · 伦理委员会与知情同意书(informed consent)是保障受试者权益的主要措 药物施,它们的实施要遵循赫尔辛基宣言
4 需要做哪些临床试验