1新药的临床前研究3
2新药的临床试验3
3GLP和GCP3
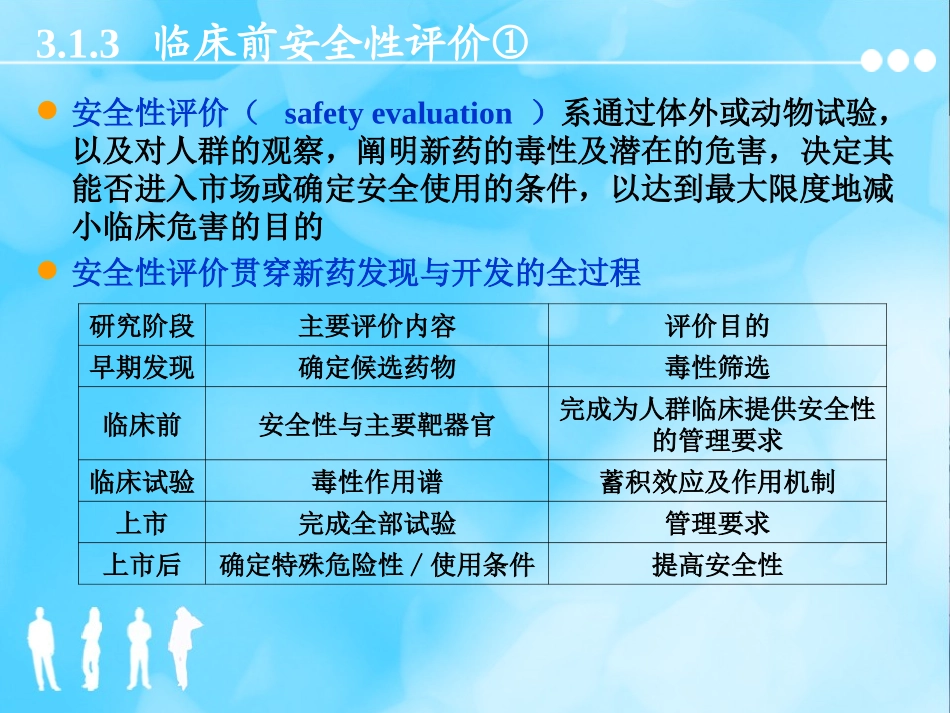
3临床前安全性评价①安全性评价(safetyevaluation)系通过体外或动物试验,以及对人群的观察,阐明新药的毒性及潜在的危害,决定其能否进入市场或确定安全使用的条件,以达到最大限度地减小临床危害的目的安全性评价贯穿新药发现与开发的全过程研究阶段主要评价内容评价目的早期发现确定候选药物毒性筛选临床前安全性与主要靶器官完成为人群临床提供安全性的管理要求临床试验毒性作用谱蓄积效应及作用机制上市完成全部试验管理要求上市后确定特殊危险性/使用条件提高安全性3
3临床前安全性评价②临床前安全性评价(即毒理学试验研究)内容:•急性毒性试验——单剂量和/或多剂量短期给药
给药剂量向一定范围增加,以确定试验化合物不产生毒性的最大剂量、发生严重毒性的剂量水平及中等毒性剂量水平•长期毒性试验——用于人体一周或以上的药物,必须要有90-180天的动物试验表明其安全性;若是用于慢性疾病治疗,必须进行一年或更长的动物试验•生殖毒性试验——包括抚养和交配行为、胚胎早期、早产和产后发育、多代影响和致畸性•遗传毒性试验——测定试验化合物是否引起基因突变或引起微粒体或DNA的损伤3
3临床前安全性评价②临床前安全性评价(即毒理学试验研究)内容:•致癌试验——当一个化合物具有足够的前景进入人体临床试验时才进行•依赖性试验——分为神经药理学试验、躯体依赖性试验和精神依赖性试验•毒代动力学试验——运用药代动力学的原理和方法,定量地研究毒性剂量下,药物在动物体内的吸收、分布、代谢、排泄过程及特点•刺激性、过敏性和溶血性试验——药物制剂经眼、耳、鼻、口腔、呼吸道、关节腔、皮肤、直肠、阴道、静脉、动脉、肌肉、皮下、静脉旁和鞘内等非口服途径给药,对用药局部产生的毒性(如刺激性和过敏性等)及对全身产生的毒性(如过敏性和溶血性