遇到长QT综合征病人,医生怎么办
长QT综合征(LongQTSyndrome,LQTS)亦称QT间期延长综合征,是一种心室复极延迟的疾病,表现为心电图(ECG)上QT间期延长(图1),这种QT间期延长可能是先天的也可能是获得的
受累患者处于晕厥、癫痫、恶性心律失常的危险之中,常导致猝死
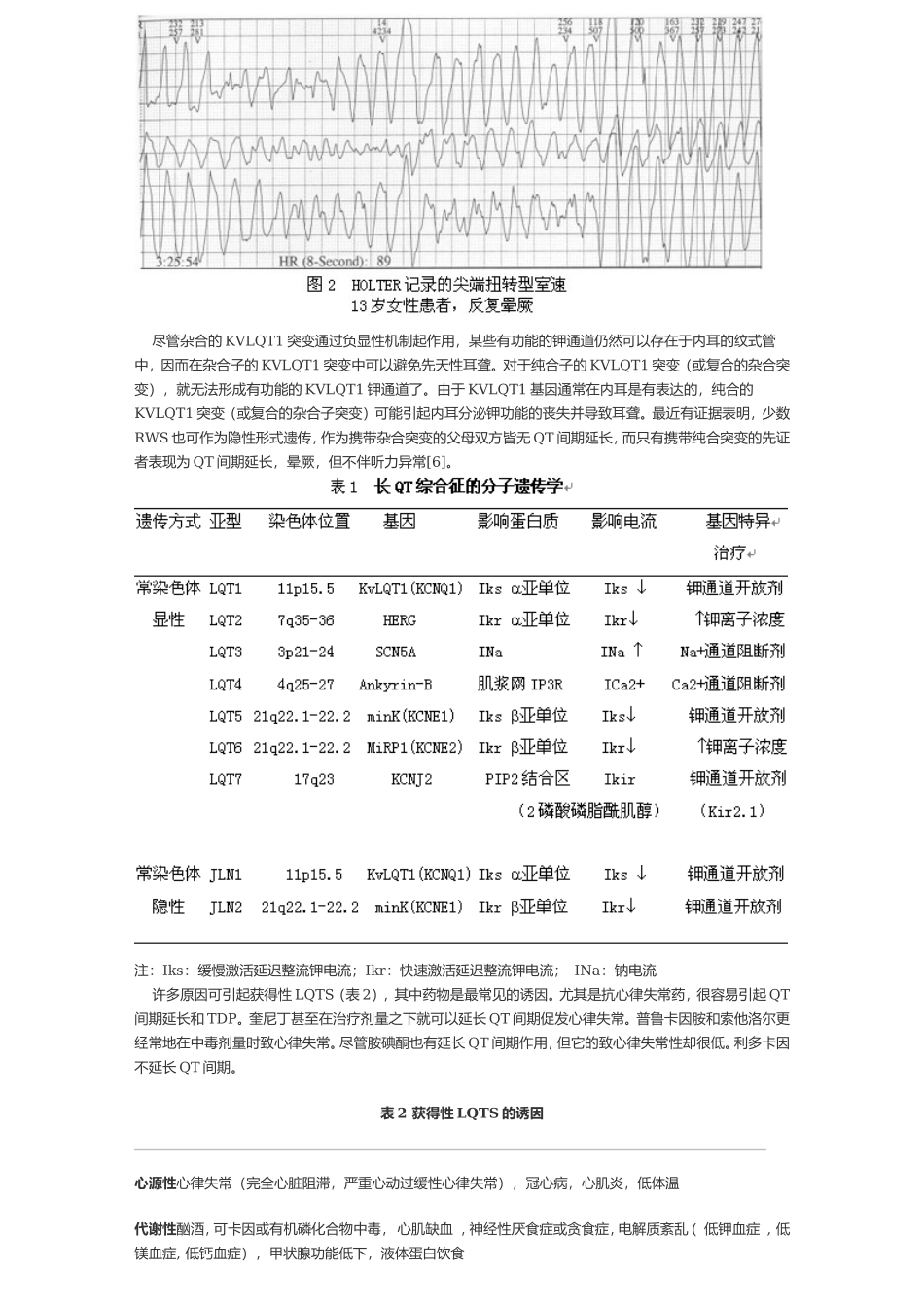
这种特征性的心律失常是一种多形性室性心动过速,称做尖端扭转型室速(Torsadedepointes,TdP),由法国的Dessertenne首先于1966年提出
该名词很好地描述了QRS波群主轴沿基线上下翻转变化的现象(图2)
在心律失常发作期间,心脏输出严重受损,常导致晕厥或癫痫样发作
尽管这些事件多数是自限性的,但这种心律失常还是有导致室颤和猝死的潜在可能
由于潜在的致命性后果,及时识别LQTS患者就显得极为重要
1LQTS的病因学LQTS可以是先天性的,也可以是获得性的
1957年Jervell和Lange-Nielsen报告了遗传性LQT的第一种形式,后被称做Jervell-Lange-Nielsen(JLN)综合征
他们描述了一个挪威家庭,6个孩子中有4个患先天性耳聋,QT延长和反复晕厥,其中3个发生猝死
父母健康,无晕厥或其它病史
1963年,Romano等和Ward独立报告了听力正常但有QT延长和晕厥的病例
这种更常见的遗传性LQT形式被称做Romano-Ward(RWS)综合征
从90年代初开始,一系列里程碑式的研究奠定了遗传性LQTS的分子遗传学基础
LQTS是第一种已证实由编码离子通道的基因突变而引起的心律失常
RWS至今已有7个基因亚型,而JLN有2个(表1)
通常RWS为常染色体显性遗传,父母只要有一方携带异常基因既可传给后代
JLN为常染色体隐性遗传,父母双方必须都携带有异常突变才会使子女受累
一般JLN患者症状更严重,猝死的几率也更高
也有报道偶发病例由新发基因突