进行性肌营养不良诊疗指南概述进行性肌营养不良(Progressivemusculardystrophy)是--组以骨骼肌进行性无力萎缩为主要临床表现的异质性基因缺陷性疾病
可伴有中枢神经系统、心脏、骨骼、呼吸及胃肠道受累
不同类型起病时间、进展速度、受累范围、严重程度差异很大
遗传方式分为 X连锁隐性遗传、常染色体显性遗传、常染色体隐性遗传等
目前已发现的致病基因达数十种
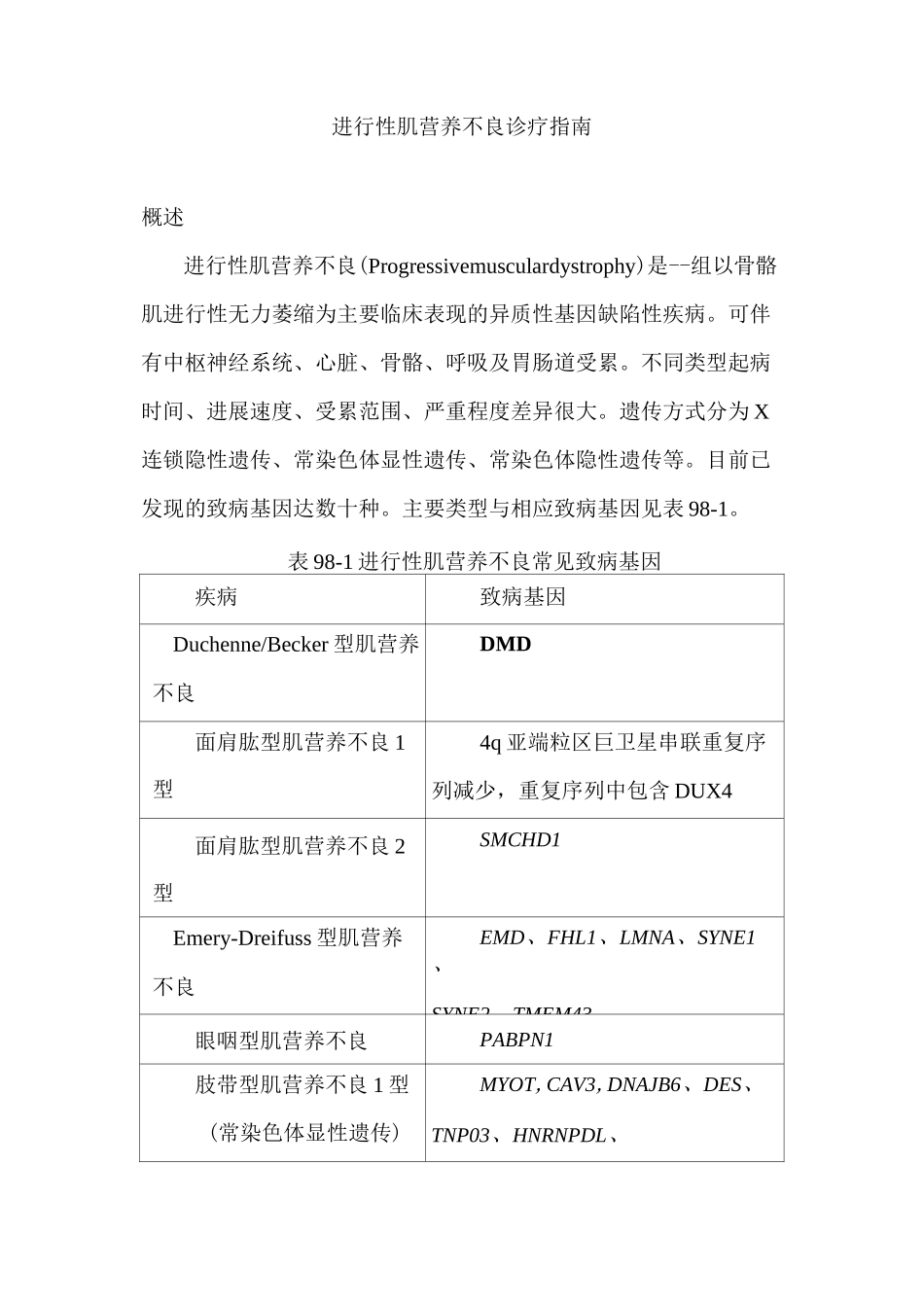
主要类型与相应致病基因见表 98-1
表 98-1 进行性肌营养不良常见致病基因疾病致病基因Duchenne/Becker 型肌营养不良DMD面肩肱型肌营养不良 1型4q 亚端粒区巨卫星串联重复序列减少,重复序列中包含 DUX4面肩肱型肌营养不良 2型SMCHD1Emery-Dreifuss 型肌营养不良EMD、FHL1、LMNA、SYNE1、SYNE2、TMEM43眼咽型肌营养不良PABPN1肢带型肌营养不良 1 型(常染色体显性遗传)MYOT,CAV3,DNAJB6、DES、TNP03、HNRNPDL、肢带型肌营养不良 2 型(常染色体隐性遗传)CAPN3、DYSF、SGCG、SGCA,SGCB、SGCD、TCAP、TRIM32、TTN、ANO5、PLEC、TRAPPC11、TOR1AIP1、LIMS2、BVES、P0GLUT1、B4GAT1Dystroglycan 糖基化相关肌营养不良P0MT1、P0MT2、P0MGNT1、FKTN、FKRP、LARGE]、ISPD、P0MGNT2、DAG1、TMEM5、B3GALNT2、P0MK、B3GNT1、GMPPB先天性肌营养不良LAMA2、C0L6A1、C0L6A2、C0L6A3、C0L12A1、SELEN0N、ITGA7、CHKB、TRIP4、INPP5K本组疾病虽有一定共性,但不同疾病诊治原则有很大不同
下面以代表性疾病 Duchenne/Becker