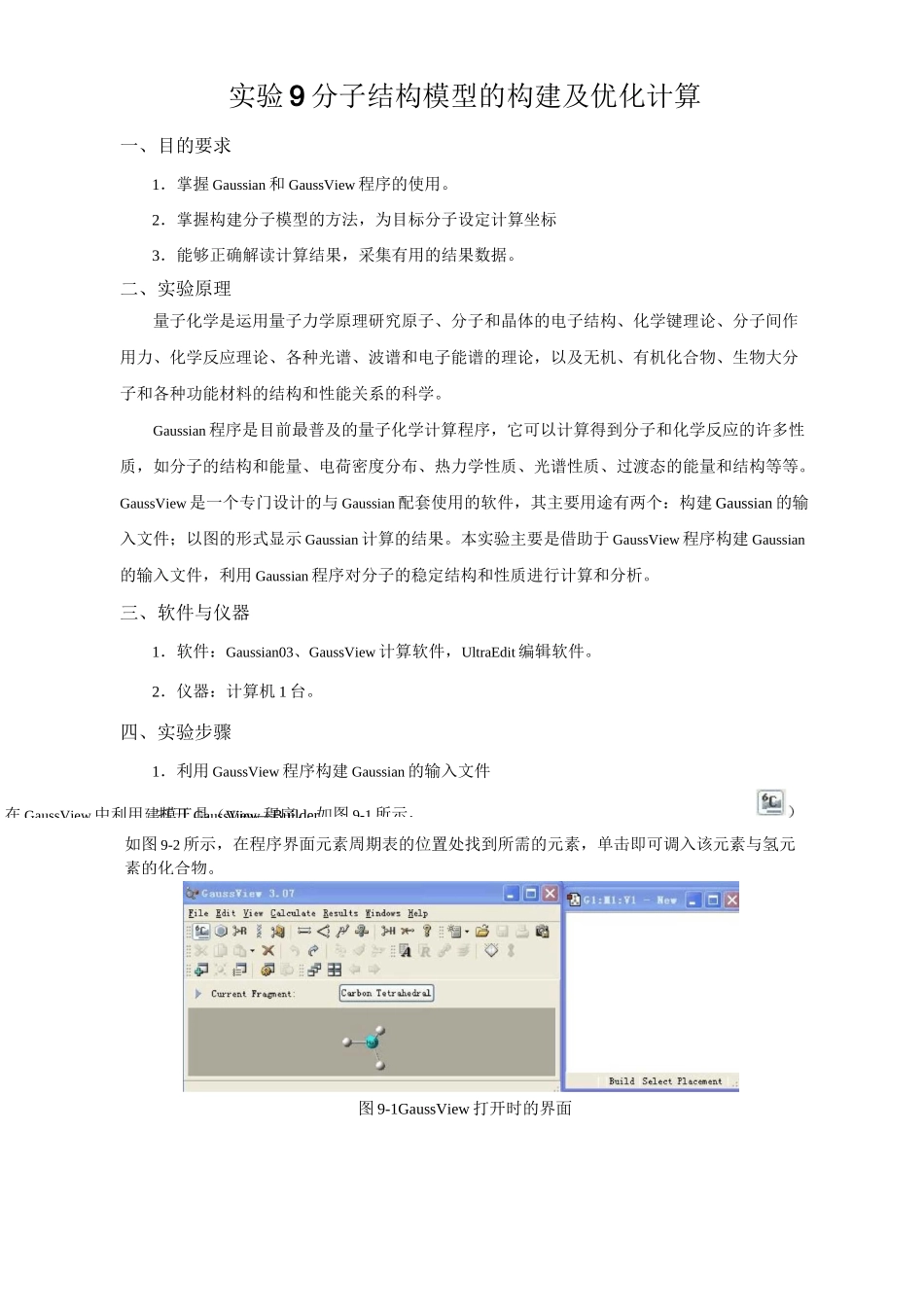
打开GaussView程序,如图9-1所示,在GaussView中利用建模工具(View—Builder实验9分子结构模型的构建及优化计算一、目的要求1.掌握Gaussian和GaussView程序的使用
2.掌握构建分子模型的方法,为目标分子设定计算坐标3.能够正确解读计算结果,采集有用的结果数据
二、实验原理量子化学是运用量子力学原理研究原子、分子和晶体的电子结构、化学键理论、分子间作用力、化学反应理论、各种光谱、波谱和电子能谱的理论,以及无机、有机化合物、生物大分子和各种功能材料的结构和性能关系的科学
Gaussian程序是目前最普及的量子化学计算程序,它可以计算得到分子和化学反应的许多性质,如分子的结构和能量、电荷密度分布、热力学性质、光谱性质、过渡态的能量和结构等等
GaussView是一个专门设计的与Gaussian配套使用的软件,其主要用途有两个:构建Gaussian的输入文件;以图的形式显示Gaussian计算的结果
本实验主要是借助于GaussView程序构建Gaussian的输入文件,利用Gaussian程序对分子的稳定结构和性质进行计算和分析
三、软件与仪器1.软件:Gaussian03、GaussView计算软件,UltraEdit编辑软件
2.仪器:计算机1台
四、实验步骤1.利用GaussView程序构建Gaussian的输入文件图9-1GaussView打开时的界面如图9-2所示,在程序界面元素周期表的位置处找到所需的元素,单击即可调入该元素与氢元素的化合物
图9-2点击Builder及双击图标°主』后出现的元素周期表窗口图若要构建像乙烷这样的链状分子,需要先点击工具栏中的按钮「,常见的链状分子就显示在新打开的窗口中,如图9-3所示
图9-3常见链状官能团窗口图若要构建像苯、萘等环状结构的分子结构,需要双击工具栏中的按钮,常见的环状有机分子就显