NCBI 在线Blast 的图文说明 Posted on 14 五月 2009 by 柳城 ,阅读 4,824 简洁版 Blast( Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA 数据库中进行相似性比较的分析工具
BLAST 程序能迅速与公开数据库进行相似性序列比较
BLAST结果中的得分是对一种对相似性的统计说明
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列
Blast 中常用的程序介绍: 1、 BLASTP 是蛋白序列到蛋白库中的一种查询
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对
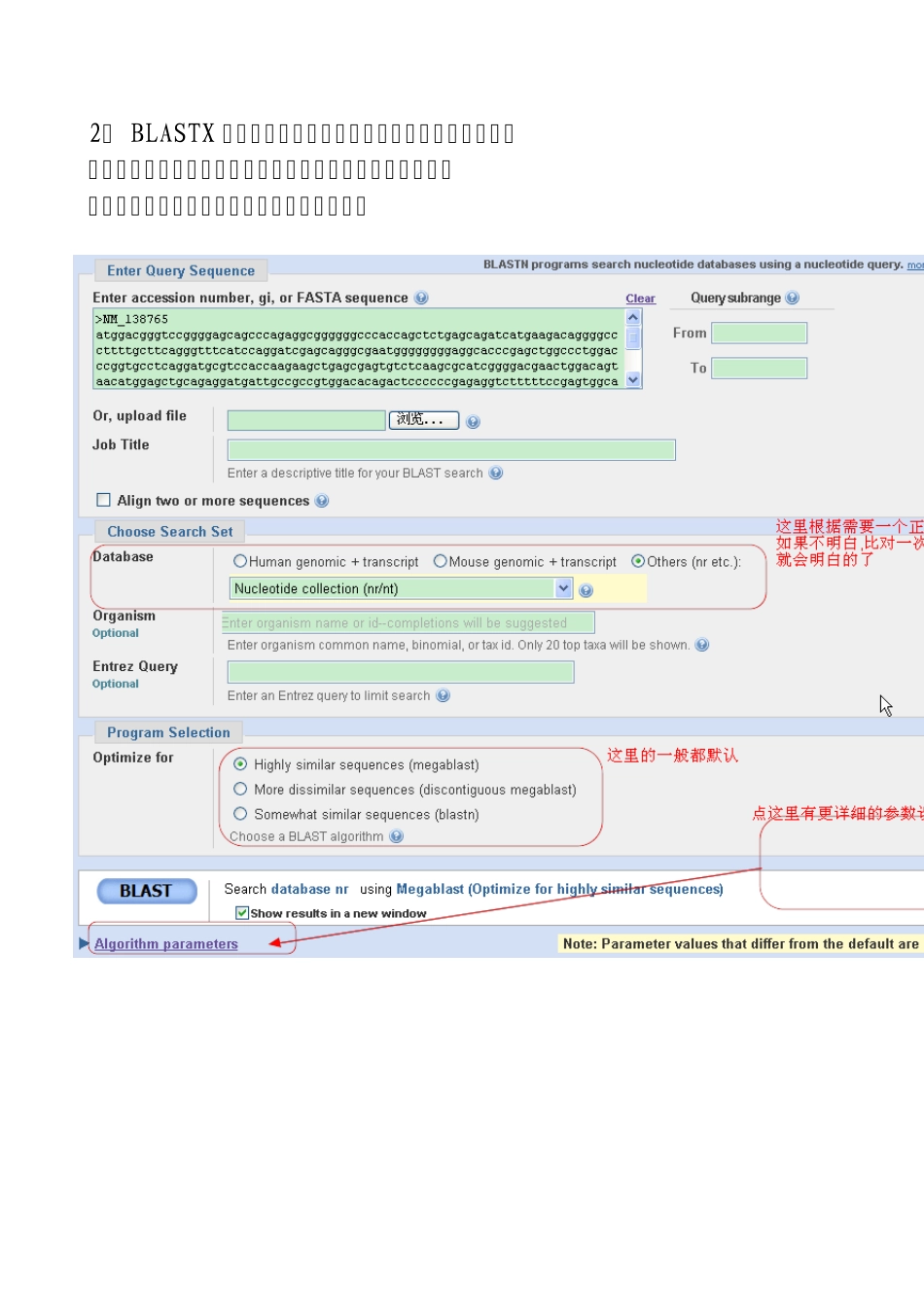
2、 BLASTX 是核酸序列到蛋白库中的一种查询
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对
3、 BLASTN 是核酸序列到核酸库中的一种查询
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对
4、 TBLASTN 是蛋白序列到核酸库中的一种查询
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对
5、 TBLASTX 是核酸序列到核酸库中的一种查询
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6 条可能的蛋白序列),这样每次比对会产生36种比对阵列
6, blast 结果的描述区域
注意分值与E 值
分值越大越靠前了,E 值越小也是这样
7, blast 结果的详细比对结果
注意比对到的序列长度
评价一个blast 结果的标准主要有三项,E 值(Expect),一致性 (Identities),缺失或插入(Gaps)
加上长度的话,就有四个标准了
如图中显示,比对到的序列长度为1405,看Identities 这一值,才匹配到1344bp,而输入的序列长