郎格罕细胞组织细胞增生症郎格罕细胞组织细胞增生症LangerhansCellHistiocytosisLangerhansCellHistiocytosis,,LCHLCH18681868年,年,PaulLangerhansPaulLangerhans最早发现最早发现了上皮树突状细胞(了上皮树突状细胞(epidermaldendriticepidermaldendriticcellscells),现在以他的名字命名为),现在以他的名字命名为LangerhansCellLangerhansCell(郎格罕细胞)
(郎格罕细胞)
由于对这类细胞已有了深入的认识并对由于对这类细胞已有了深入的认识并对它们进行了分类,故用”朗格罕细胞组它们进行了分类,故用”朗格罕细胞组织细胞增生症织细胞增生症(Langerhanscell(Langerhanscellhistiocytosis)”histiocytosis)”代替原来的“组织细胞代替原来的“组织细胞增生症增生症X(histiocytosisX)”X(histiocytosisX)”
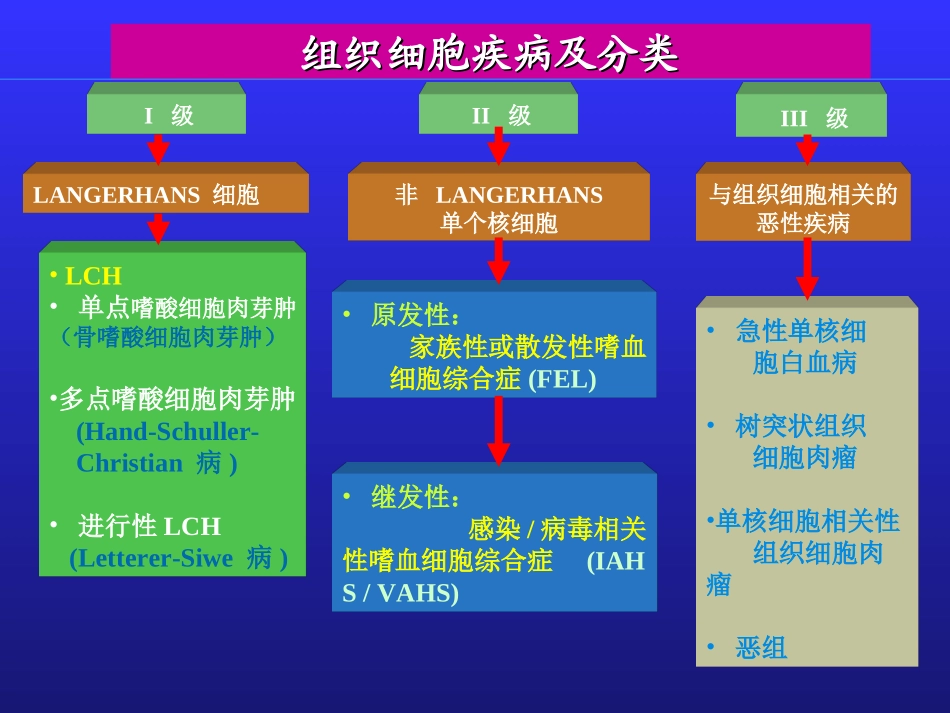
I级LANGERHANS细胞II级非LANGERHANS单个核细胞•原发性:家族性或散发性嗜血细胞综合症(FEL)•继发性:感染/病毒相关性嗜血细胞综合症(IAHS/VAHS)III级与组织细胞相关的恶性疾病•急性单核细胞白血病•树突状组织细胞肉瘤•单核细胞相关性组织细胞肉瘤•恶组组织细胞疾病及分类组织细胞疾病及分类•LCH•单点嗜酸细胞肉芽肿(骨嗜酸细胞肉芽肿)•多点嗜酸细胞肉芽肿(Hand-Schuller-Christian病)•进行性LCH(Letterer-Siwe病)临床表现与分型临床表现与分型((一)勒一)勒--雪病雪病((Letterer-Siwedisease,LSLetterer-Siwedisease,LS))1