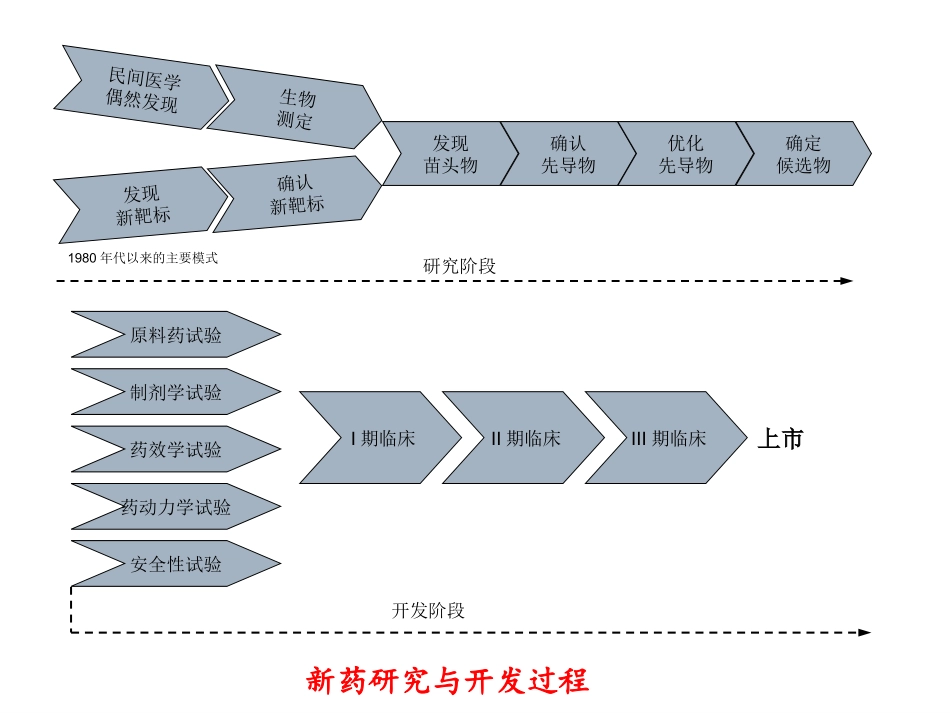
药物的分子设计策略药物的分子设计策略(3)(3)::先导化先导化合物的质量与优化合物的质量与优化一、概述新药创制过程:将非药的活性化合物向成药转化,满足安全、有效、稳定和质量可控的要求
生物学:活性评价模型和评价方法化学:发现苗头化合物(hit)和(或)先导化合物(lead),优化结构,确定一批有成药前景的物质,即候选药物(drugcandidate)按照药政法规对候选药物进行系统的临床前研究,经审批后进入临床I期、II期和III期研究,最终经批准上市应用这是一条研究开发链,确定候选药物是个重要环节民间医学偶然发现生物测定发现新靶标确认新靶标发现苗头物确认先导物优化先导物确定候选物研究阶段1980年代以来的主要模式原料药试验制剂学试验药效学试验药动力学试验安全性试验I期临床II期临床III期临床上市开发阶段新药研究与开发过程新药研发各个环节的价值贡献度先导物的发现与优化约占价值链10%,时程约3-5年,但决定了后面90%的命运优化先导物并确定候选药物对于新药创制的成败至关重要;候选药物的质量取决于先导物的优劣和优化准则,发现和确定高质量先导物是重要的起点
从苗头化合物到先导物苗头化合物(hit):对特定靶标或作用环节具有初步活性的化合物
苗头化合物的发现途径:理性设计(基于受体或配体结构和机制的分子设计)随机筛选(天然产物和高通量筛选化合物库)基于片段的筛选(仪器分析和分子模拟相结合的技术)HajdukPJ.etal
NatRevDrugDiscov,2007,6:211—219.SiegalG.etal
DrugDiscovToday,2007,12:1032—1039苗头化合物未必都能进入研究阶段,因为固有的缺陷不能发展成先导物活性表现为非特异作用活性表现为非特异作用药代动力学不合理药代动力学不合理物化性质差物化性质差毒副作用大毒副作