欧洲共同体: European Communities (EC)
欧洲联盟: European Union (EU) ,简称欧盟
人用药品注册技术标准国际协调会:ICH 欧盟 GMP附录1 无菌药品的生产文件历史上一版本 2003年5月30日发布自2003年9月生效修订了洁净间级别表,培养基模拟试验、生物负荷监测和冻干小瓶轧盖的指南
2005年11月至2007年12月生效并替代前版2009年3月1日注:冻干瓶轧盖的条款自 2010年3月1日开始实施
原则为降低微生物、微粒和热原污染的风险,无菌药品的生产应有各种特殊要求
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法和规程进行
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验
注:本指南没有对微粒、浮游菌和表面微生物等测试方法详细进行阐述,可参阅欧洲标准或国际标准(CEN/ISO)及药典资料
无菌药品的生产必须在洁净区内进行,人员和(或)设备以及物料必须通过缓冲进入洁净区
洁净区应当保持适当的洁净度,洁净区的送风须经具有一定过滤效率过滤器的过滤
原料配制、产品加工和灌装等不同操作必须在洁净去内彼此分开的单独区域内进行
生产工艺可分为两类:一类是最终灭菌工艺;第二类是部分或全部工序为无菌操作的工艺
应按所需环境的特点确定无菌产品的洁净级别
每一步生产操作都应达到适当的动态洁净度,以尽可能降低产品(或原料)被微粒或微生物污染
洁净区的设计必须符合相应的“静态”标准,以达到“动态”的洁净要求
“静态”是指安装已经完成并已运行,但没有操作人员在场的状态
“动态”是指生产设施按预定的工艺模式运行并有规定数量的操作人员进行现场操作的状态
应确定每一洁净室或每组洁净间的“动态”及“静态”标准
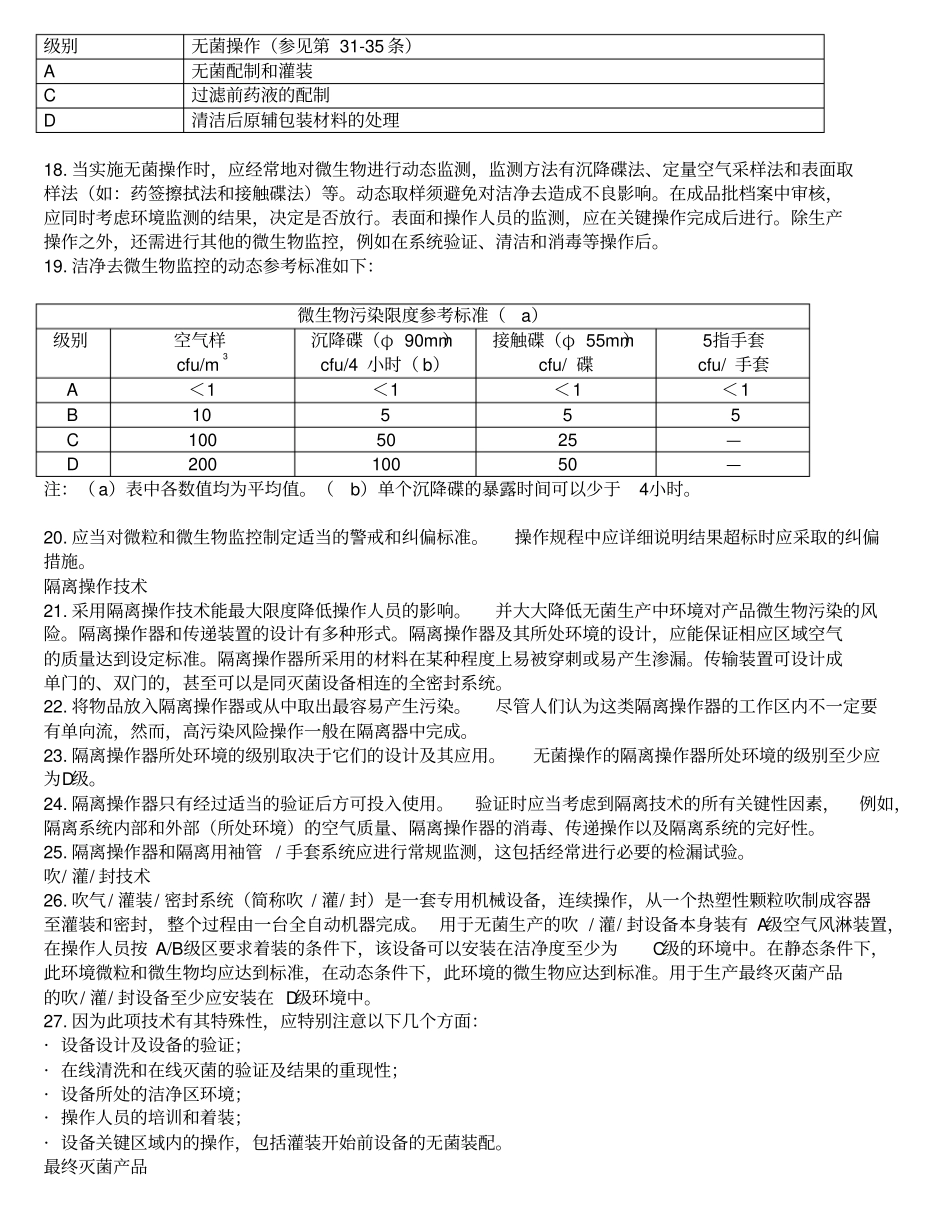
无菌药品生产所需的洁净区